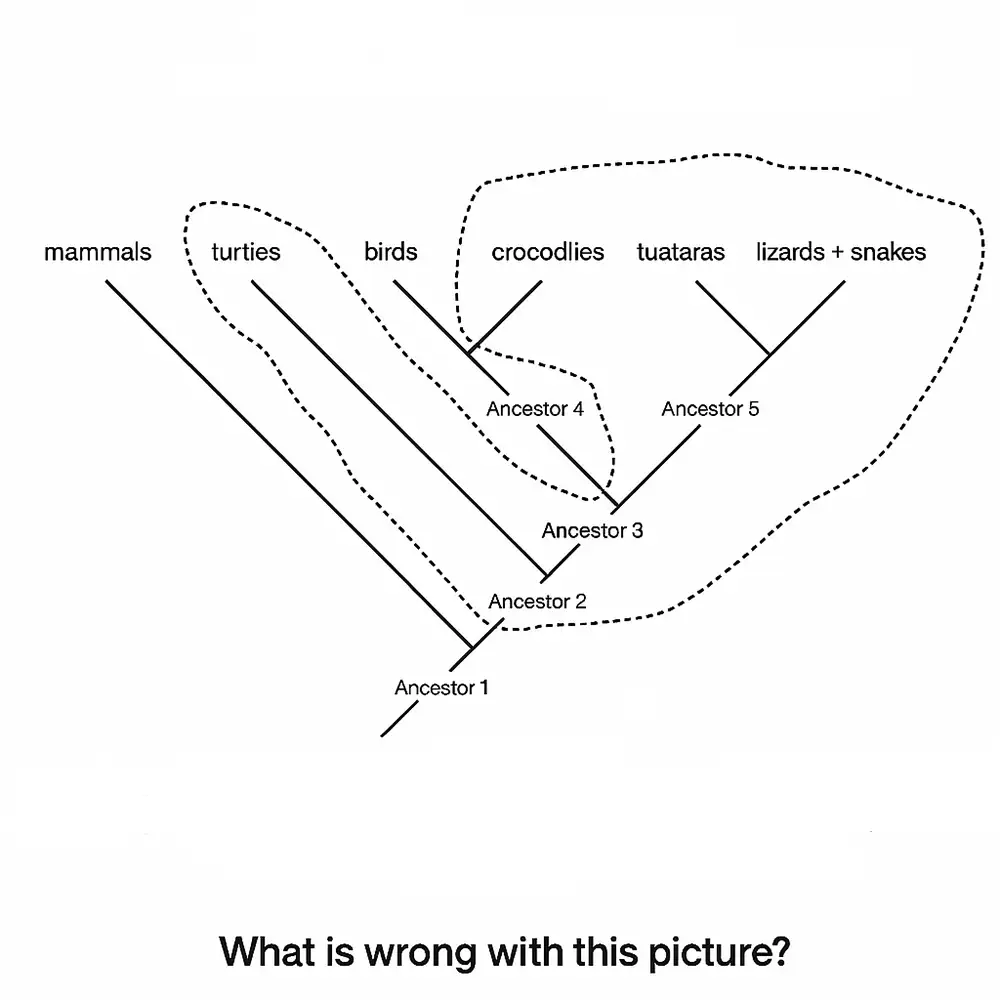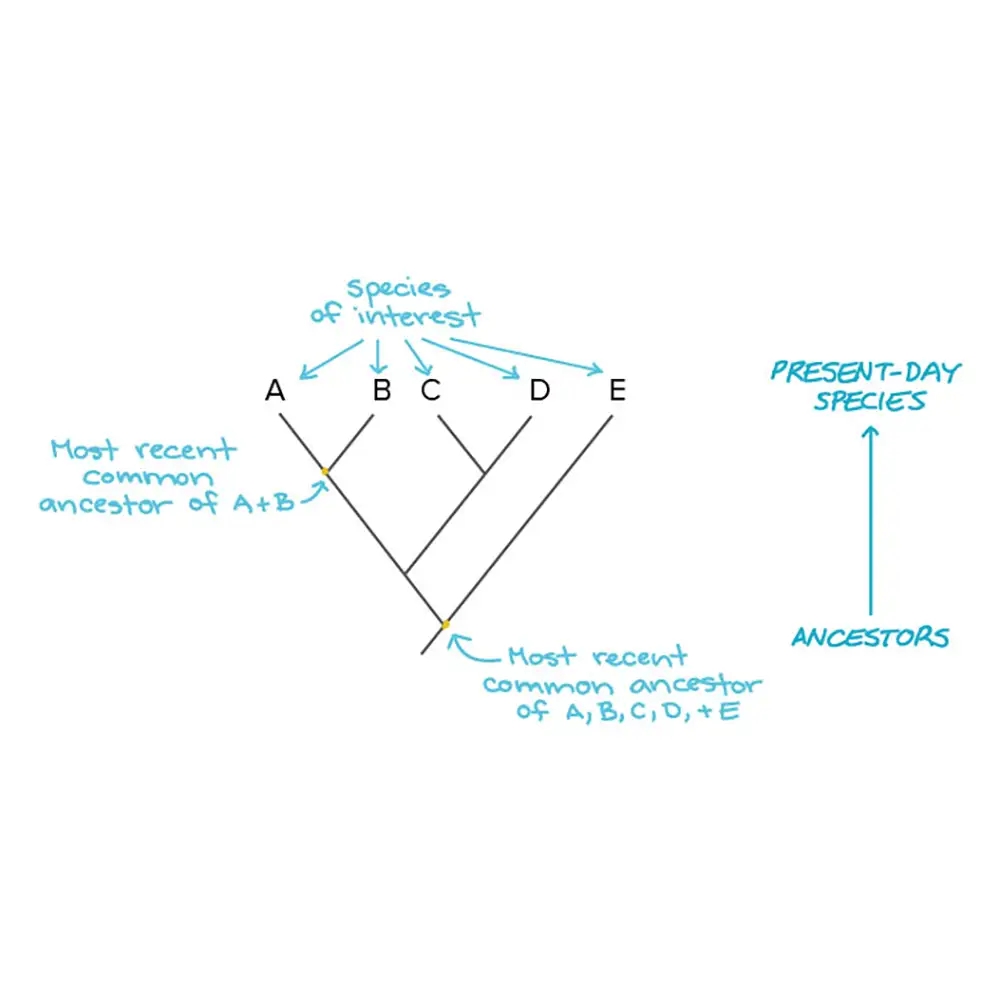
روش های ساخت درخت تبارزایی
بخش ۱: مقدمه
درختهای فیلوژنتیک ابزار مرکزی در درک روابط تکاملی بین موجودات زنده هستند. واژهٔ فیلوژنتیک از دو کلمهٔ یونانی phylon به معنی «قبیله» یا «نژاد» و genetikos به معنی «منشأ» یا «نسل» گرفته شده است. اساساً، فیلوژنتیک تلاش میکند مسیرهای تاریخیای را بازسازی کند که گونهها و واحدهای تاکسونومی دیگر از نیای مشترک اجداد خود تکامل یافتهاند. درختهای فیلوژنتیک، که نمودارهایی از این روابط هستند، چارچوب مفهومیای برای درک الگوهای شاخهای تکامل، زمانبندی رویدادهای انشعاب، و میزان نزدیکی موجودات در سطوح مختلف تاکسونومی ارائه میدهند.
اهمیت تحلیل فیلوژنتیک فراتر از زیستشناسی کلاسیک است. در زیستشناسی تکاملی، درختهای فیلوژنتیک به فهم رویدادهای گونهزایی، تشعشعات تطبیقی و تکامل ویژگیهای پیچیده کمک میکنند. در ژنومیکس، این درختها برای تحلیلهای مقایسهای، شناسایی ژنها و شبکههای تنظیمی محافظتشده و عناصر عملکردی در گونهها کاربرد دارند. در پزشکی و اپیدمیولوژی، درختهای فیلوژنتیک برای پیگیری تکامل پاتوژنها، ردیابی منابع شیوع بیماری و مطالعهٔ گسترش مقاومت ضد میکروبی استفاده میشوند. زیستشناسی حفاظتی از این درختها برای شناسایی شاخههای تکاملی متمایز و اولویتبندی گونهها برای حفاظت بهره میبرد، و اکولوژی از آنها برای درک چگونگی شکلگیری جوامع، همتکامل و تکامل ویژگیها استفاده میکند.
تاریخچهٔ تحلیل فیلوژنتیک به چندین قرن قبل بازمیگردد. سیستمهای طبقهبندی اولیه عمدتاً فنوتیپیک بودند و موجودات را بر اساس شباهت کلی گروهبندی میکردند، بدون توجه صریح به سلف مشترک. کارلوس لینه (Linnaeus) در قرن هجدهم سیستم نامگذاری دوتایی را ایجاد کرد و چارچوبی استاندارد برای نامگذاری گونهها فراهم نمود. هرچند سیستم لینهای سلسلهمراتبی بود، ولی بهطور صریح روابط تکاملی را نشان نمیداد. مفهوم نزول مشترک (Common Descent) که توسط چارلز داروین در کتاب منشأ گونهها (۱۸۵۹) معرفی شد، زیستشناسی را متحول کرد و تأکید کرد که گروهبندیهای تاکسونومیک باید تاریخچهٔ تبارشناسی را بازتاب دهند.
اولین درختهای فیلوژنتیک رسمی بر اساس ویژگیهای مورفولوژیکی، مانند ساختارهای آناتومیکی، اسکلت و ویژگیهای رشد و نمو طراحی شدند. با پیشرفت زیستشناسی مولکولی در اواسط قرن بیستم، دنبالههای مولکولی—DNA، RNA و پروتئینها—به منبع اصلی اطلاعات فیلوژنتیکی تبدیل شدند. دادههای مولکولی امکان مقایسههای عینی و کمّی بیشتر را فراهم میکنند و وضوح بالاتری ایجاد کرده و بازسازی روابط تکاملی عمیق را ممکن میسازند که دادههای مورفولوژیک به تنهایی قادر به آن نبودند.
فیلوژنتیک مدرن روشهای محاسباتی، آماری و بیوانفورماتیکی را برای مدیریت حجم عظیم دادههای تولیدشده توسط تکنولوژیهای توالییابی پرسرعت ترکیب میکند. الگوریتمها و مدلهای پیشرفته به پژوهشگران اجازه میدهند درختهای فیلوژنتیک قوی و قابل اعتماد بسازند، فرضیههای تکاملی را آزمایش کنند و میزان اطمینان از روابط بازسازیشده را بسنجند. به این ترتیب، درختهای فیلوژنتیک از نمودارهای ساده به ابزارهای علمی پیچیدهای تبدیل شدهاند که پایهٔ زیستشناسی و تحقیقات پزشکی معاصر را تشکیل میدهند.
بخش ۲: مفاهیم بنیادی
برای درک روشهای ساخت درختهای فیلوژنتیک، لازم است مفاهیم بنیادی در زیستشناسی تکاملی، تاکسونومی و سیستماتیک به خوبی شناخته شوند. این بخش اصطلاحات اصلی، انواع درختها و اصول پایه لازم برای تحلیل فیلوژنتیک را معرفی میکند.
۲.۱ تاکسونومی، سیستماتیک و فیلوژنتیک
-
تاکسونومی علمی است که به نامگذاری، توصیف و طبقهبندی موجودات زنده میپردازد. این علم دستهبندیهای سلسلهمراتبی مانند گونه، جنس، خانواده و راسته را تعیین میکند. تاکسونومی چارچوب استانداردی برای سازماندهی تنوع زیستی فراهم میآورد، ولی به خودی خود روابط تکاملی را نشان نمیدهد.
-
سیستماتیک شامل تاکسونومی است، ولی تمرکز آن بر درک روابط تکاملی بین موجودات میباشد. سیستماتیک از دادههای مورفولوژیکی، مولکولی، اکولوژیکی و رفتاری برای بازسازی تاریخچهٔ حیات استفاده میکند.
-
فیلوژنتیک شاخهای از سیستماتیک است که بهطور خاص به استنتاج روابط تکاملی میپردازد. درختهای فیلوژنتیک خروجی اصلی تحلیلهای فیلوژنتیک هستند و فرضیههایی دربارهٔ اجداد و تبار موجودات ارائه میکنند.
۲.۲ انواع درختهای فیلوژنتیک
درختهای فیلوژنتیک میتوانند چند شکل مختلف داشته باشند که هر کدام تفسیر متفاوتی ارائه میکنند:
درختهای ریشهدار (Rooted) در مقابل درختهای بدون ریشه (Unrooted):
-
درختهای ریشهدار جهت زمان تکاملی را نشان میدهند و موقعیت آخرین جد مشترک (MRCA) تمام تاکسونها را مشخص میکنند.
-
درختهای بدون ریشه فقط روابط بین تاکسونها را نشان میدهند، بدون اینکه خط تبار یا جهت زمانی مشخص شود و معمولاً زمانی استفاده میشوند که ریشه معلوم نیست.
کلادوگرامها، فیلوگرامها و دندروگرامها:
-
کلادوگرامها ترتیب شاخهها را نشان میدهند، ولی اطلاعاتی درباره طول شاخه یا میزان تغییر تکاملی ارائه نمیکنند.
-
فیلوگرامها طول شاخهها را بر اساس میزان تغییر تکاملی (مثلاً جایگزینی نوکلئوتیدها در توالیهای مولکولی) مقیاس میکنند.
-
دندروگرامها نمودارهای سلسلهمراتبی هستند که معمولاً در تحلیلهای فنوتیپیک برای خوشهبندی موجودات بر اساس شباهت کلی استفاده میشوند و ممکن است روابط تکاملی واقعی را نشان ندهند.
۲.۳ کاراکترها در تحلیل فیلوژنتیک
استنتاج فیلوژنتیکی بر مقایسه کاراکترها بین تاکسونها استوار است. کاراکترها میتوانند شامل موارد زیر باشند:
-
مورفولوژیکی: ساختارهای آناتومیکی، ویژگیهای جنینی یا دیگر صفات قابل مشاهده.
-
مولکولی: توالیهای DNA، RNA یا پروتئین. در فیلوژنتیک مدرن، کاراکترهای مولکولی ترجیح داده میشوند، زیرا دادههای زیاد و قابل مقایسه فراهم میکنند و امکان مدلسازی آماری فرآیندهای تکاملی را میدهند.
هر کاراکتر میتواند در حالتهای مختلف وجود داشته باشد (مثلاً حضور یا غیاب، یا نوکلئوتیدهای خاص در DNA). تشخیص صحیح کاراکترهای همولوگ—یعنی آنهایی که از جد مشترک به ارث رسیدهاند—اهمیت زیادی دارد، در مقابل کاراکترهای آنالوگ که بهطور مستقل و در نتیجه تکامل همگرا ایجاد شدهاند.
۲.۴ همولوژی در مقابل آنالوژی
-
همولوژی (Homology): شباهت به دلیل تبار مشترک. مثالها شامل اندامهای جلویی پستانداران یا ژنهای محافظتشده در مهرهداران است.
-
آنالوژی (Analogy یا Homoplasy): شباهت ناشی از تکامل همگرا یا موازی، یعنی ویژگیهای مشابه در شاخههای نامرتبط بهطور مستقل تکامل یافتهاند. تشخیص همولوژی از آنالوژی بسیار مهم است، زیرا همولوژی اشتباه میتواند تحلیل فیلوژنتیک را گمراه کند.
۲.۵ روشهای مبتنی بر فاصله در مقابل مبتنی بر کاراکتر
روشهای فیلوژنتیک را میتوان به دو دسته اصلی تقسیم کرد:
روشهای مبتنی بر فاصله (Distance-based):
-
فاصلهٔ تکاملی جفتبهجفت بین تاکسونها را محاسبه میکنند.
-
درختهایی تولید میکنند که بهترین بازتابدهندهٔ فاصلهها باشند.
-
مثالها: UPGMA، Neighbor-Joining
روشهای مبتنی بر کاراکتر (Character-based):
-
تغییرات هر کاراکتر را بین تاکسونها بررسی میکنند.
-
به دنبال درختهایی هستند که توزیع مشاهدهشدهٔ کاراکترها را طبق معیارهای مشخص توضیح دهند (مانند پارسیمونی، بیشینه درستنمایی یا احتمال بیزی).
-
مثالها: Maximum Parsimony، Maximum Likelihood، Bayesian Inference
این تفاوتهای مفهومی انتخاب روشهای مناسب در ساخت درختهای فیلوژنتیک را تعیین میکنند. هر رویکرد دارای مزایا، محدودیتها و فرضیات خاص خود است که در بخشهای بعدی بهطور مفصل بررسی خواهد شد.
بخش ۳: دادهها برای تحلیل فیلوژنتیک
تحلیل فیلوژنتیک بر دادههای دقیق و اطلاعاتی تکیه دارد. کیفیت، کمیت و نوع دادهها مستقیماً بر قابلیت اعتماد روابط تکاملی بازسازیشده تأثیر میگذارند. در گذشته، ویژگیهای مورفولوژیکی منبع اصلی اطلاعات فیلوژنتیکی بودند. با این حال، در فیلوژنتیک مدرن، به طور فزایندهای از دادههای مولکولی شامل توالیهای DNA، RNA و پروتئین و همچنین دادههای ژنومی و ترنسکریپتومی استفاده میشود. این بخش مروری دقیق بر انواع مختلف دادهها که در ساخت درختهای فیلوژنتیک استفاده میشوند ارائه میدهد و به چالشها و ملاحظات انتخاب و آمادهسازی این دادهها برای تحلیل میپردازد.
۳.۱ دادههای مورفولوژیکی
دادههای مورفولوژیکی به ویژگیهای قابل مشاهدهٔ آناتومیکی و فیزیولوژیکی موجودات اطلاق میشود. این نوع دادهها تاریخچهٔ بازسازی فیلوژنتیک را تشکیل دادهاند.
۳.۱.۱ انواع کاراکترهای مورفولوژیکی
-
مورفولوژی خارجی: اندازه و شکل بدن، رنگ، ساختار برگ یا گل در گیاهان، یا ساختار اندامها و ارگانها در حیوانات.
-
مورفولوژی داخلی: ساختارهای اسکلتی، سیستمهای ارگانها، عضلات یا آرایشهای عصبی.
-
ویژگیهای جنینی: الگوهای رشد و نمو، مانند بخشبندی، تشکیل لایههای جنینی و توسعهٔ رویانی (Ontogeny).
-
ویژگیهای رفتاری: رفتارهای جفتگیری، استراتژیهای تغذیه یا ساختارهای اجتماعی (در برخی مطالعات فیلوژنتیک بهطور انتخابی استفاده میشوند).
۳.۱.۲ مزایای دادههای مورفولوژیکی
-
مستقیماً قابل مشاهده هستند و اغلب به تجهیزات تخصصی نیاز ندارند.
-
برای گونههای منقرضشده (فسیلها) که داده مولکولی در دسترس نیست، ضروری هستند.
-
میتوانند بینشهایی دربارهٔ سازگاریهای عملکردی و جایگاههای اکولوژیکی ارائه دهند.
۳.۱.۳ محدودیتها
-
همگرایی و هموپلازی (Homoplasy) میتواند بازسازی درخت را گمراه کند (مثلاً بال در پرندگان و خفاشها).
-
کدگذاری ذهنی کاراکترها: تعیین اینکه کدام ویژگیها امتیازدهی شوند و چگونه حالتهای کاراکتر اختصاص یابند، میتواند سوگیری ایجاد کند.
-
وضوح محدود برای روابط تکاملی عمیق، به ویژه در میان میکروارگانیسمها.
۳.۲ دادههای توالی مولکولی
دادههای مولکولی انقلابی در فیلوژنتیک ایجاد کردهاند و اطلاعات کمی و با وضوح بالا ارائه میدهند که میتوان آنها را با روشهای آماری و محاسباتی تحلیل کرد.
۳.۲.۱ توالیهای DNA
-
DNA هستهای: اطلاعات سراسر ژنوم را ارائه میدهد و برای استنتاج روابط در سطح گونهها و مطالعهٔ تکامل خانوادههای ژنی مفید است. چالشها شامل بازترکیبی و رویدادهای تکثیر ژن است.
-
DNA میتوکندریایی (mtDNA): معمولاً از مادر به ارث میرسد و نسبتاً سریع تکامل مییابد. برای مطالعات جمعیت و فیلوژئوشیمی رایج است.
-
DNA کلروپلاست (cpDNA): به دلیل ماهیت محافظتشده و وراثت مادری در فیلوژنتیک گیاهی استفاده میشود.
۳.۲.۲ توالیهای RNA
-
rRNA (RNA ریبوزومی): مناطق بسیار محافظتشده اجازه میدهند توالیها بین تاکسونهای دور مرتبط همتراز (Alignment) شوند، در حالی که مناطق متغیر وضوح در سطوح تاکسونومی پایینتر را فراهم میکنند.
-
mRNA (RNA پیامرسان): نشاندهنده ژنهای بیانشده است و برای مطالعات ترنسکریپتومیک و تکامل عملکردی کاربرد دارد.
۳.۲.۳ توالیهای پروتئینی
-
توالیهای آمینواسیدی استخراجشده از مناطق کدکننده میتوانند بین تاکسونها همتراز و مقایسه شوند.
-
پروتئینها نسبت به DNA کندتر تکامل مییابند، که برای مطالعه روابط تکاملی دور مزیت دارد.
-
افزایش کدونها و محدودیتهای عملکردی باید هنگام تفسیر تفاوتهای توالی در نظر گرفته شود.
۳.۳ دادههای ژنومی و ترنسکریپتومی
تکنولوژیهای توالییابی پرسرعت امکان استفاده از دادههای ژنومی و ترنسکریپتومی بزرگمقیاس را فراهم کردهاند و وضوح بیسابقهای برای تحلیل فیلوژنتیک ارائه میدهند.
۳.۳.۱ توالییابی ژنوم کامل
-
نمای کامل ساختار ژنتیکی یک موجود را ارائه میدهد.
-
امکان فیلوژنومیکس (Phylogenomics) را فراهم میکند، جایی که هزاران ژن همزمان تحلیل میشوند.
-
به شناسایی تکثیر ژن، انتقال ژن افقی و بازآراییهای ساختاری کمک میکند.
۳.۳.۲ ترنسکریپتومیکس
-
RNA-seq امکان تحلیل الگوهای بیان ژنها در گونهها را فراهم میکند.
-
برای حل روابط بین تاکسونهای نزدیک یا فهم تکامل تطبیقی مفید است.
-
نیاز به نرمالسازی دقیق و تصحیح برای سطوح مختلف بیان دارد.
۳.۴ نشانگرهای مولکولی
نشانگرهای مولکولی توالیهای خاص DNA هستند که بهعنوان نمایندهای از میزان انحراف تکاملی استفاده میشوند.
۳.۴.۱ انواع رایج
-
RFLP (Restriction Fragment Length Polymorphisms): تفاوت اندازهٔ قطعات DNA پس از هضم آنزیمی.
-
SSR / میکروستلیتها (Simple Sequence Repeats): توالیهای کوتاه تکرارشونده، بسیار پلیمورفیک و مفید در مطالعات جمعیتی.
-
SNP (Single Nucleotide Polymorphisms): تغییرات تک نوکلئوتیدی، فراوان و مفید برای فیلوژنتیک با وضوح بالا.
-
AFLP (Amplified Fragment Length Polymorphisms): شناسایی تغییرات در چندین لوکوس به طور همزمان.
۳.۴.۲ معیارهای انتخاب
-
نشانگرها باید همولوگ بین تاکسونها باشند تا مقایسه معنیدار باشد.
-
سطح تغییرپذیری باید با مقیاس تکاملی مورد نظر مطابقت داشته باشد (نشانگرهای سریع برای انشعابات اخیر، نشانگرهای محافظتشده برای فیلوژنیهای عمیق).
۳.۵ چالشها در انتخاب و آمادهسازی دادهها
استنتاج دقیق فیلوژنتیک به انتخاب دقیق دادهها، پیشپردازش و کنترل کیفیت وابسته است. نکات کلیدی شامل:
-
ارزیابی همولوژی: فقط کاراکترها یا توالیهای همولوگ باید مقایسه شوند. دادههای غیرهمولوگ نویز ایجاد میکنند.
-
کیفیت همترازی (Alignment): برای توالیهای مولکولی، همترازی چندتایی حیاتی است. همترازی نادرست میتواند منجر به ساختار درختی اشتباه شود.
-
دادههای ناقص: دادههای ناکامل، فاصلهها یا توالیهای مبهم قابلیت اعتماد درخت را کاهش میدهند. استراتژیها شامل تخمین مقادیر گمشده، حذف سایتهای مشکلدار یا اصلاح مبتنی بر مدل است.
-
تغییر نرخ تکاملی بین سایتها: برخی کاراکترها سریعتر از بقیه تکامل مییابند. در نظر گرفتن ناهمگنی نرخها از سوگیری در ساخت درخت جلوگیری میکند.
-
نمونهگیری تاکسونها: گنجاندن مجموعهای نماینده از تاکسونها باعث کاهش جذب شاخههای طولانی و بهبود وضوح میشود. نمونهگیری کم یا جانبدارانه ممکن است روابط تکاملی را تحریف کند.
بخش ۴: همترازی توالی بهعنوان پیشنیاز
همترازی توالی (Sequence Alignment) یک گام اساسی در تحلیل فیلوژنتیک مولکولی است. دقت درخت فیلوژنتیک تا حد زیادی به کیفیت همترازی وابسته است، زیرا مشخص میکند که کدام موقعیتهای نوکلئوتید یا آمینواسید در بین تاکسونها همولوگ محسوب میشوند. همترازی نادرست میتواند منجر به استنتاج اشتباه روابط تکاملی شود، در حالی که همترازی با کیفیت بالا اجازه میدهد مناطق محافظتشده و متغیر، الگوهای جایگزینی و Indelها (درج یا حذف) بهطور قابل اعتماد شناسایی شوند. این بخش مروری جامع بر همترازی توالی، انواع آن، الگوریتمها، چالشها و ارتباط آن با ساخت درخت فیلوژنتیک ارائه میدهد.
۴.۱ تعریف و هدف همترازی توالی
همترازی توالی فرآیند چینش توالیهای DNA، RNA یا پروتئین است تا مناطق مشابه شناسایی شوند، که ممکن است نشاندهنده همولوژی یا تبار مشترک باشند. هدف این است که توالیها به گونهای همتراز شوند که موقعیتهای مرتبط تکاملی (نوکلئوتید یا آمینواسید) در یک ستون ماتریس قرار گیرند و مقایسهٔ معنیدار بین تاکسونها امکانپذیر شود.
اهداف همترازی توالی در فیلوژنتیک شامل موارد زیر است:
-
شناسایی موقعیتهای همولوگ برای مقایسه کاراکترها.
-
تشخیص تغییرات تکاملی مانند جایگزینی، درج یا حذف.
-
برآورد انحراف توالی که اطلاعات لازم برای روشهای مبتنی بر فاصله را فراهم میکند.
-
آمادهسازی دادهها برای روشهای مبتنی بر کاراکتر، جایی که ستونهای همتراز بهعنوان کاراکتر برای Maximum Parsimony، Maximum Likelihood یا Bayesian Inference استفاده میشوند.
۴.۲ انواع همترازی توالی
همترازی توالی به دو نوع کلی تقسیم میشود:
۴.۲.۱ همترازی جفتبهجفت (Pairwise Alignment)
-
شامل همترازی دو توالی بهطور همزمان است.
-
برای تشخیص شباهت بین توالیها، شناسایی اورتوگها یا ساخت درخت راهنما برای همترازی چندتایی مفید است.
الگوریتمها:
-
الگوریتم Needleman–Wunsch (همترازی سراسری / Global Alignment): توالیها را از ابتدا تا انتها همتراز میکند و شباهت کلی را بهینه میکند. مناسب توالیهای هماندازه یا مشابه.
-
الگوریتم Smith–Waterman (همترازی محلی / Local Alignment): مناطق بیشترین شباهت در توالیها را همتراز میکند و امکان همپوشانی جزئی را میدهد. مناسب توالیهایی با طول متغیر یا شامل درج/حذف.
۴.۲.۲ همترازی چندتایی (Multiple Sequence Alignment, MSA)
-
شامل همترازی سه یا چند توالی همزمان است.
-
برای تحلیل فیلوژنتیک حیاتی است زیرا موقعیتهای محافظتشده و متغیر در بین چند تاکسون را شناسایی میکند.
الگوریتمها و ابزارها:
-
همترازی پیشرونده (Progressive Alignment): مانند ClustalW و Clustal Omega
-
توالیها را تدریجی بر اساس یک درخت راهنما همتراز میکند.
-
سریع است اما ممکن است خطاهای اولیه را در طول همترازی منتقل کند.
-
-
همترازی تکراری (Iterative Alignment): مانند MUSCLE و MAFFT
-
با همترازی مکرر توالیها یا زیرگروهها دقت را بهبود میبخشد.
-
دقت بالاتری برای توالیهای متفاوت و دور دارد.
-
-
روشهای مبتنی بر ثبات (Consistency-based): مانند T-Coffee
-
اطلاعات چندین همترازی جفتبهجفت را ترکیب میکند تا قابلیت اعتماد افزایش یابد.
-
۴.۳ امتیازدهی و بهینهسازی
همترازی توالی با سیستمهای امتیازدهی کیفیت همترازی را ارزیابی میکند:
-
امتیازدهی تطبیق / عدم تطبیق:
-
به جایگاههای یکسان یا مشابه امتیاز مثبت و به عدم تطبیقها امتیاز منفی اختصاص میدهد.
-
-
جریمه شکافها (Gap Penalties):
-
برای در نظر گرفتن درج یا حذف (Indel) اعمال میشود.
-
جریمه خطی (Linear Gap Penalty): هر شکاف هزینه یکسان دارد.
-
جریمه افاین (Affine Gap Penalty): باز شدن شکاف جریمه بالاتری نسبت به طول آن دارد، که منعکسکننده احتمال زیستی است.
-
-
ماتریسهای جایگزینی (Substitution Matrices):
-
معمولاً برای توالیهای پروتئینی کاربرد دارد.
-
مثالها: PAM، BLOSUM
-
احتمال جایگزینی آمینواسیدها را بر اساس مدلهای تکاملی اختصاص میدهند.
-
الگوریتمهای همترازی به دنبال بهینه کردن امتیاز کل هستند و تعادلی بین مطابقتها، عدم تطابقها و شکافها برقرار میکنند.
۴.۴ چالشها در همترازی توالی
-
Indelها و مناطق تکراری: درج و حذف باعث پیچیدگی همترازی میشوند، به ویژه در توالیهای دور از هم.
-
توالیهای بسیار متفاوت: با افزایش تفاوت توالیها، شناسایی همولوژی دشوارتر میشود و خطاهای همترازی میتوانند منجر به استنتاج اشتباه در روشهای Maximum Likelihood یا Bayesian شوند.
-
پیچیدگی محاسباتی: همترازی بهینه چندتایی NP-hard است. الگوریتمهای ابتکاری و پیشرونده دقت کمی را فدای قابلیت محاسباتی میکنند.
-
عدم قطعیت همترازی: برخی مناطق ممکن است ابهامآمیز همتراز شوند. ماسک کردن یا حذف مناطق بدهمتراز میتواند قابلیت اعتماد درخت را بهبود دهد.
۴.۵ اصلاح و ارزیابی کیفیت همترازی
برای اطمینان از استنتاج فیلوژنتیک دقیق، همترازیها اغلب اصلاح و ارزیابی میشوند:
-
ویرایش دستی:
-
با بازبینی بصری میتوان خطاهای آشکار را اصلاح کرد، به ویژه در مناطق محافظتشده.
-
-
اصلاح خودکار:
-
ابزارهایی مانند Gblocks یا TrimAl مناطق بدهمتراز یا بسیار متغیر را حذف میکنند.
-
-
بررسی ثبات:
-
مقایسه چندین روش همترازی برای اطمینان از قابلیت اعتماد. مناطق ناسازگار ممکن است حذف یا با احتیاط استفاده شوند.
-
-
امتیازدهی و ارزیابی:
-
Sum-of-pairs score یا Column score کیفیت همترازی را ارزیابی میکنند.
-
روشهای Bootstrap میتوانند اثر عدم قطعیت همترازی بر توپولوژی درخت را بررسی کنند.
-
۴.۶ همترازی توالی در تحلیل فیلوژنتیک
-
همترازی دقیق پیشنیاز تمام روشهای مولکولی فیلوژنتیک است:
-
روشهای مبتنی بر فاصله (UPGMA، Neighbor-Joining) به شباهت یا انحراف جفتبهجفت توالیها وابستهاند که از همترازی استخراج میشود.
-
روشهای مبتنی بر کاراکتر (Maximum Parsimony، Maximum Likelihood، Bayesian Inference) هر ستون همتراز را بهعنوان یک کاراکتر در نظر میگیرند و نیازمند تخصیص دقیق همولوژی هستند.
-
-
توالیهای بدهمتراز میتوانند خطاهای سیستماتیک مانند Synapomorphyهای کاذب (ویژگیهای مشترک اکتسابی) یا Homoplasyهای ساختگی ایجاد کنند و توپولوژی درخت را تحریف کنند.
-
همترازی همچنین پایهٔ انتخاب مدل در روشهای مبتنی بر Likelihood است. مدلهای جایگزینی نوکلئوتید یا آمینواسید معمولاً به الگوهای مشاهدهشده در توالیهای همتراز بستگی دارند.
بخش ۵: روشهای ساخت درخت فیلوژنتیک
ساخت درخت فیلوژنتیک فرآیندی است برای استنتاج روابط تکاملی میان موجودات یا ژنها بر اساس دادههای مقایسهای. پس از جمعآوری دادههای توالی یا مورفولوژیکی و همتراز کردن صحیح آنها، میتوان از روشهای محاسباتی مختلف برای تولید درخت استفاده کرد. این روشها را میتوان به طور کلی به سه دسته تقسیم کرد:
-
روشهای مبتنی بر فاصله (Distance-Based Methods)
-
روشهای مبتنی بر کاراکتر (Character-Based Methods)
-
روشهای پیشرفته آماری یا روشهای اجماعی (Consensus or Advanced Statistical Approaches)
هر روش بر اصول و فروض خاص خود متکی است و از نظر پیچیدگی محاسباتی، دقت و تناسب با انواع دادهها متفاوت است. درک این روشها برای انتخاب روش مناسب و تفسیر درختهای حاصل ضروری است.
۵.۱ روشهای مبتنی بر فاصله (Distance-Based Methods)
روشهای مبتنی بر فاصله با محاسبه ماتریس فاصله جفتبهجفت شروع میشوند که میزان انحراف تکاملی بین همه تاکسونها را نشان میدهد. این روشها دادههای پیچیده توالی یا کاراکترها را به یک شاخص واحد اختلاف تبدیل میکنند و سپس تاکسونها را بر اساس شباهت کلی خوشهبندی میکنند.
-
مزیت: سرعت بالا و مصرف محاسباتی کمتر نسبت به روشهای مبتنی بر کاراکتر، مناسب برای دادههای بزرگ.
-
معایب: تکیه بر فاصلههای خلاصه شده ممکن است اطلاعات تکاملی را بیش از حد سادهسازی کند و دقت را در دادههای پیچیده کاهش دهد.
۵.۱.۱ روش گروهبندی زوجی بدون وزن با میانگین حسابی (UPGMA)
-
UPGMA یکی از سادهترین و قدیمیترین روشهای مبتنی بر فاصله است.
-
از خوشهبندی سلسلهمراتبی استفاده میکند و به صورت تکراری زوج تاکسونها یا خوشههایی با کمترین فاصله را به هم میپیوندد.
-
فرض میکند که تمام خطوط تکاملی با نرخ ثابتی تکامل مییابند (ساعت مولکولی).
-
نتیجه، درخت ریشهدار اولترامتریک است که گرهها از ریشه با فاصله برابر قرار دارند.
-
مزیت: محاسباتی کارآمد و پیادهسازی ساده.
-
محدودیت: اگر نرخ تکامل بین شاخهها متفاوت باشد، دقت کاهش مییابد.
۵.۱.۲ روش همسایهیابی (Neighbor-Joining, NJ)
-
روش NJ انعطافپذیرتر و رایجتر است و به ساعت مولکولی نیاز ندارد.
-
با شناسایی زوج تاکسونها یا خوشههایی که طول شاخه کل را حداقل میکنند، درخت را به صورت درخت بدون ریشه میسازد.
-
مزیت: ترکیب کارایی محاسباتی با دقت بالاتر برای دادههای با نرخهای تکاملی متفاوت.
-
مناسب برای دادههای بزرگ که روشهای Maximum Likelihood یا Parsimony در آنها زمانبر است.
-
محدودیت: هنوز به دقت ماتریس فاصله وابسته است؛ خطا در فاصلهها میتواند توپولوژی را تحت تأثیر قرار دهد.
۵.۱.۳ روش فیتچ–مارگولیاش (Fitch–Margoliash)
-
Fitch–Margoliash فاصلهها را بر اساس واریانس تخمینی آنها وزندهی میکند.
-
با دادن وزن بیشتر به فاصلههای قابل اعتمادتر، دقت ساخت درخت افزایش مییابد، به ویژه وقتی که میزان انحراف توالیها بین تاکسونها متفاوت است.
-
مانند سایر روشهای مبتنی بر فاصله، پیچیدگی محاسباتی کمتر نسبت به روشهای مبتنی بر کاراکتر دارد، اما اطلاعات جزئی کاراکترها ممکن است در فرآیند خلاصهسازی فاصلهها از دست برود.
۵.۲ روشهای مبتنی بر کاراکتر (Character-Based Methods)
روشهای مبتنی بر کاراکتر هر کاراکتر یا موقعیت توالی را مستقیماً تحلیل میکنند و آنها را به فاصله جفتبهجفت خلاصه نمیکنند.
-
این روشها تمام توپولوژیهای ممکن درخت را ارزیابی میکنند تا درختی که بهترین توضیح برای توزیع کاراکترها ارائه میدهد را شناسایی کنند.
-
مزیت: حفظ کامل اطلاعات دادهها، دقت بالاتر از روشهای مبتنی بر فاصله.
-
معایب: پیچیدگی محاسباتی بالاتر، به ویژه برای تعداد زیاد تاکسونها یا کاراکترها.
۵.۲.۱ حداقل تغییر (Maximum Parsimony, MP)
-
بر اصل تیغ اوکام (Occam’s razor) مبتنی است: درختی که کمترین تغییر تکاملی لازم برای توضیح دادهها را داشته باشد، بهترین است.
-
هر ستون همتراز (نوکلئوتید، آمینواسید یا کاراکتر مورفولوژیک) به عنوان یک کاراکتر جداگانه در نظر گرفته میشود.
-
الگوریتمهایی مانند Fitch یا Wagner Parsimony برای ارزیابی درختها استفاده میشوند.
-
مزیت: ساده و مؤثر برای دادههایی با کمترین Homoplasy.
-
محدودیتها: حساسیت به جذب شاخه طولانی (Long-branch attraction) و افزایش پیچیدگی محاسباتی بهطور نمایی با افزایش تعداد تاکسونها، نیازمند استراتژیهای جستجوی ابتکاری (Heuristic).
۵.۲.۲ حداکثر احتمال (Maximum Likelihood, ML)
-
از چارچوب آماری استفاده میکند تا احتمال دادههای مشاهده شده با توجه به یک درخت و مدل تکامل را ارزیابی کند.
-
ML احتمال تغییرات کاراکتر در هر سایت را بر اساس مدلهای جایگزینی مانند:
-
Jukes–Cantor (JC69)
-
Kimura دو پارامتری (K2P)
-
Hasegawa–Kishino–Yano (HKY85)
-
General Time Reversible (GTR)
محاسبه میکند.
-
-
به دنبال توپولوژی درختی است که احتمال دادهها را بیشینه کند.
-
مزیتها: دقت بالا، امکان در نظر گرفتن تغییر نرخ بین سایتها، فرکانسهای پایه نامساوی و سایر واقعیتهای بیولوژیکی.
-
Bootstrapping برای ارزیابی قابلیت اعتماد شاخهها استفاده میشود.
۵.۲.۳ استنتاج بیزی (Bayesian Inference)
-
یک رویکرد احتمالاتی در فیلوژنتیک است که اطلاعات پیشین درباره پارامترهای تکاملی را با دادههای مشاهدهشده ترکیب میکند تا احتمال پسین توپولوژیهای درخت را محاسبه کند.
-
با الگوریتمهایی مانند Markov Chain Monte Carlo (MCMC)، توزیع درختهای محتمل نمونهگیری میشود، نه انتخاب یک درخت منفرد.
-
نرمافزارهایی مانند MrBayes و BEAST پیادهسازی این روش هستند.
-
مزیت: پشتیبانی آماری قوی، امکان در نظر گرفتن مدلهای پیچیده تکامل توالی.
-
چالشها: پیچیدگی محاسباتی بالا و نیاز به انتخاب دقیق توزیعهای پیشین برای جلوگیری از سوگیری.
۵.۳ روشهای دیگر
علاوه بر روشهای کلاسیک، روشهای دیگری برای حل چالشهای خاص فیلوژنتیک توسعه یافتهاند:
-
روشهای اجماعی (Consensus Methods): ترکیب چندین درخت برای تولید یک درخت خلاصه، مانند Strict Consensus یا Majority-Rule Consensus.
-
روشهای چهارتایی (Quartet Methods): روابط بین تمام زیرمجموعههای چهار تاکسون را بررسی و سپس آنها را به درخت کلان assemble میکنند.
-
روشهای مبتنی بر کوآلِسِنت (Coalescent-Based Methods): مدلسازی تکامل درخت ژنی در درخت گونهها، با در نظر گرفتن تغییرات ناتمام نسلها و سایر ناسازگاریها.
-
روشهای سوپردرخت و سوپرماتریس (Supertree & Supermatrix): ادغام اطلاعات از چندین مجموعه داده یا خانواده ژنی برای ساخت فیلوژنی جامع، کاربردی در فیلوژنومیکس.
-
روشهای یادگیری ماشین (Machine Learning): استفاده از هوش مصنوعی و یادگیری عمیق برای استنتاج روابط تکاملی از الگوهای توالی یا ژنومی، مناسب برای تحلیل سریع دادههای حجیم.
۵.۴ انتخاب روش فیلوژنتیک
انتخاب روش مناسب به عوامل زیر بستگی دارد:
-
نوع و حجم دادهها: دادههای بزرگ ممکن است روشهای مبتنی بر فاصله یا ML ابتکاری را ترجیح دهند، در حالی که دادههای کوچک و باکیفیت مناسب Parsimony یا ML کامل هستند.
-
ناهمگنی نرخ تکاملی: روشهای ML و Bayesian نرخهای متغیر را میتوانند مدل کنند، در حالی که UPGMA فرض ساعت مولکولی سختگیرانه دارد.
-
منابع محاسباتی: Bayesian و ML ممکن است نیازمند کامپیوترهای پرقدرت برای دادههای بزرگ باشند.
-
اهداف پژوهشی: مطالعات اکتشافی ممکن است از روشهای سریع مبتنی بر فاصله استفاده کنند، در حالی که مطالعاتی که پشتیبانی دقیق شاخهها میخواهند، از ML یا Bayesian همراه با Bootstrapping یا احتمال پسین استفاده میکنند.
بخش ۶: ابزارها و نرمافزارهای محاسباتی
ساخت درختهای فیلوژنتیک همزمان با پیشرفتهای زیستشناسی محاسباتی تکامل یافته است. تحلیلهای مدرن فیلوژنتیک معمولاً شامل دادههای حجیم، مدلهای تکاملی پیچیده و روشهای آماری پیشرفته هستند، بنابراین ابزارهای محاسباتی ضروری به نظر میرسند. این نرمافزارها نه تنها الگوریتمهای مختلف ساخت درخت را پیادهسازی میکنند، بلکه امکاناتی برای همترازی توالیها، انتخاب مدل، بوتاسترپینگ، تصویرسازی و تفسیر نتایج نیز ارائه میدهند. این بخش مروری بر نرمافزارهای پرکاربرد فیلوژنتیک، ویژگیها، نقاط قوت، محدودیتها و نکات عملی برای پژوهشگران ارائه میدهد.
۶.۱ MEGA (Molecular Evolutionary Genetics Analysis)
-
MEGA یکی از پرکاربردترین نرمافزارها برای تحلیل فیلوژنتیک و مطالعات تکامل مولکولی است.
-
محیط یکپارچهای برای همترازی توالیها، محاسبه فاصلههای تکاملی، ساخت درخت و ارزیابی آماری فراهم میکند.
-
از روشهای مختلف ساخت درخت مانند UPGMA، Neighbor-Joining، Maximum Parsimony و Maximum Likelihood پشتیبانی میکند و امکان بوتاسترپینگ برای ارزیابی قابلیت اعتماد شاخهها را دارد.
-
مزایا: رابط گرافیکی و آموزشهای گسترده، مناسب برای مبتدیان و اهداف آموزشی.
-
ابزارهای اضافی: آزمون فرضیات تکاملی، برآورد زمان واگرایی، تحلیل الگوهای جایگزینی نوکلئوتید یا آمینواسید.
-
محدودیت: مدیریت دادههای ژنومی بسیار بزرگ ممکن است محدود باشد، به ویژه نسبت به نرمافزارهای خط فرمان بهینهشده برای محاسبات پرقدرت.
۶.۲ PHYLIP (Phylogeny Inference Package)
-
PHYLIP توسط Joseph Felsenstein توسعه یافته و یکی از مجموعه نرمافزارهای پیشگام در فیلوژنتیک است.
-
طیف وسیعی از روشهای ساخت درخت شامل روشهای مبتنی بر فاصله، پارسیمونی و حداکثر احتمال (Likelihood) را پیادهسازی میکند.
-
مزیتها:
-
ساختار ماژولار که امکان اجرای برنامههای جداگانه برای مراحل مختلف مانند محاسبه فاصله، جستجوی درخت و بوتاسترپینگ را میدهد.
-
شفافیت الگوریتمها و مستندات دقیق، مناسب برای مطالعات تحقیقاتی دقیق.
-
-
محدودیتها:
-
رابط خط فرمان و طراحی نسبتاً قدیمی، ممکن است برای کاربران جدید دشوار باشد.
-
تصویرسازی درختها معمولاً نیازمند نرمافزارهای مکمل است.
-
-
با وجود محدودیتها، PHYLIP همچنان ابزار قابل اعتماد برای مطالعات عمیق فیلوژنتیک است، به ویژه همراه با بستههای تصویرسازی مکمل.
۶.۳ RAxML (Randomized Axelerated Maximum Likelihood)
-
RAxML ابزار تخصصی برای تحلیل فیلوژنتیک با روش Maximum Likelihood است که برای دادههای حجیم و محاسبات پرقدرت بهینه شده است.
-
مدلهای پیچیده تکامل نوکلئوتید، آمینواسید و کدون را پیادهسازی میکند و از بوتاسترپ سریع و تحلیلهای تقسیمبندیشده برای چندین ژن پشتیبانی میکند.
-
کاربرد اصلی: فیلوژنومیکس، تحلیل هزاران ژن یا کل ژنومها.
-
مزیت: سرعت و مقیاسپذیری بالا برای مسائل فیلوژنتیک پیچیده و بزرگ.
-
محدودیت: نیازمند آشنایی با محیطهای محاسباتی خط فرمان برای ادغام در خطوط پردازشی خودکار.
۶.۴ IQ-TREE
-
IQ-TREE نرمافزار مدرن Maximum Likelihood است که دقت، سرعت و سهولت استفاده را ترکیب میکند.
-
ویژگیها: انتخاب خودکار مدل، بوتاسترپینگ فوق سریع، پشتیبانی از دادههای تقسیمبندیشده.
-
مزیت: تحلیل سریع و کارآمد دادههای بزرگ و متنوع.
-
پشتیبانی از طیف گستردهای از مدلهای جایگزینی و ارائه آزمونهای آماری برای ارزیابی توپولوژی درخت و پشتیبانی شاخهها.
-
کاربرد: پژوهشگران پیشرفته و مبتدیان فیلوژنتیک.
-
ویژگی کلیدی: قابلیت تحلیل دادههای مقیاس ژنومی با سرعت و دقت بالا.
۶.۵ نرمافزارهای فیلوژنتیک بیزی: MrBayes و BEAST
-
استنتاج بیزی (Bayesian Inference) نیازمند ابزارهایی است که بتوانند شبیهسازی MCMC برای محاسبه احتمال پسین توپولوژیهای درخت انجام دهند.
-
MrBayes:
-
استفاده از مدلهای پیچیده جایگزینی نوکلئوتید یا آمینواسید، در نظر گرفتن ناهمگنی نرخها و تحلیل دادههای تقسیمبندیشده.
-
خروجی شامل توزیع درختها است که میتوان درختهای اجماعی و احتمالات پسین استخراج کرد.
-
-
BEAST:
-
تخصص در فیلوژنیهای زمانبندی شده، برآورد زمان واگرایی و دینامیک جمعیت.
-
کاربرد گسترده در زیستشناسی تکاملی، اپیدمیولوژی و فیلوژئوگرافی.
-
-
محدودیت: مصرف بالای محاسباتی، اما ارائه دقت آماری بینظیر و انعطاف بالا برای تحلیل فیلوژنتیک بیزی.
۶.۶ ابزارهای آنلاین و تصویرسازی
-
علاوه بر نرمافزارهای مستقل، سرویسها و پلتفرمهای آنلاین برای تحلیل و تصویرسازی درخت وجود دارند.
-
مثالها:
-
PhyML: ساخت درخت Maximum Likelihood از طریق وب، بدون نیاز به نصب محلی.
-
ETE Toolkit و iTOL (Interactive Tree Of Life): امکانات تصویرسازی پیشرفته، حاشیهنویسی، رنگآمیزی و تعامل با درختها.
-
-
مزیت: دسترسی آسان، همکاری و ارائه نتایج، به ویژه برای پژوهشگرانی که دسترسی به منابع محاسباتی پرقدرت ندارند.
۶.۷ نکات عملی
انتخاب نرمافزار فیلوژنتیک نیازمند توجه به عوامل زیر است:
-
حجم و پیچیدگی دادهها: دادههای ژنومی بزرگ نیاز به ابزارهای بهینهشده برای محاسبات سریع مانند RAxML یا IQ-TREE دارند، دادههای کوچکتر با MEGA یا PHYLIP قابل مدیریتاند.
-
نیازمندیهای روش: تحلیلهای بیزی نیازمند MrBayes یا BEAST هستند، در حالی که روشهای مبتنی بر فاصله یا پارسیمونی با MEGA یا PHYLIP قابل انجاماند.
-
منابع محاسباتی: ML و Bayesian نیازمند منابع محاسباتی بالا و احتمالاً دسترسی به خوشههای پردازشی یا محیطهای محاسبات پرقدرت.
-
تبحر کاربر: رابط گرافیکی مانند MEGA برای مبتدیان مناسب است، ابزارهای خط فرمان انعطاف و اتوماسیون بالاتری دارند اما نیازمند دانش محاسباتی هستند.
-
تصویرسازی و تفسیر: تصویرسازی دقیق درخت برای ارتباط نتایج پژوهشی حیاتی است، بنابراین ابزارهایی مانند iTOL یا ETE Toolkit مکمل ارزشمندی برای نرمافزارهای استنتاج درخت محسوب میشوند.
بخش ۷: ارزیابی درختهای فیلوژنتیک
ساخت یک درخت فیلوژنتیک تنها بخشی از فرایند تحلیل است. به همان اندازه مهم است که اعتبار، مقاومت و اهمیت آماری درخت ارزیابی شود. استنتاج فیلوژنتیک شامل چندین فرض، انتخاب مدل و منابع بالقوه خطا است که همگی میتوانند توپولوژی و طول شاخههای درخت را تحت تأثیر قرار دهند.
ارزیابی درختهای فیلوژنتیک تضمین میکند که روابط استنتاجشده بازتابدهنده تاریخچه تکاملی واقعی هستند و نه نتیجه خطاهای داده، همترازی یا روش تحلیل. این بخش تکنیکهای اصلی برای سنجش قابلیت اعتماد درخت را بررسی میکند، از جمله بوتاسترپینگ، جکنایفینگ، احتمالات پسین و استراتژیهای تصویرسازی.
۷.۱ بوتاسترپینگ (Bootstrapping)
-
بوتاسترپینگ یکی از رایجترین روشها برای ارزیابی قابلیت اعتماد درختهای فیلوژنتیک است.
-
توسط Felsenstein در سال ۱۹۸۵ معرفی شد و شامل نمونهبرداری مجدد از مجموعه داده اصلی با جایگزینی است تا چندین شبهنمونه (Pseudo-Replicate) ایجاد شود.
-
برای هر شبهنمونه، یک درخت با همان روش فیلوژنتیک ساخته میشود.
-
مقدار پشتیبانی بوتاسترپ برای هر کلاد، درصد تکرار آن کلاد در میان مجموعه شبهنمونههاست.
-
مثال: مقدار بوتاسترپ ۹۰٪ یعنی کلاد در ۹۰٪ مجموعههای بازنمونهگیریشده ظاهر شده است، نشاندهنده پشتیبانی قوی است.
-
-
قابل استفاده برای روشهای مبتنی بر فاصله و مبتنی بر کاراکتر، به ویژه در Maximum Parsimony و Maximum Likelihood.
-
نکته مهم: مقادیر بوتاسترپ میتوانند تحت تأثیر تعداد کاراکترها، طول توالی و توپولوژی کلی درخت قرار گیرند و باید به عنوان برآوردی از قابلیت اعتماد تعبیر شوند، نه اطمینان مطلق.
۷.۲ جکنایفینگ (Jackknifing)
-
جکنایفینگ از نظر مفهومی شبیه بوتاسترپینگ است، اما شامل نمونهبرداری بدون جایگزینی میشود و معمولاً با حذف بخشی از دادهها در هر تکرار انجام میشود.
-
برای هر مجموعه داده کاهشیافته، درخت ساخته میشود و فرکانس وقوع کلادها در طول تکرارها به عنوان معیار پشتیبانی محاسبه میشود.
-
کمتر از بوتاسترپینگ استفاده میشود، اما میتواند اطلاعات مکمل ارائه دهد، به ویژه برای بررسی حساسیت توپولوژی درخت نسبت به دادههای ناقص یا متغیر.
-
هر دو روش (بوتاسترپ و جکنایف) واریانس نمونهگیری را مدنظر قرار میدهند و کمک میکنند تا پژوهشگران تشخیص دهند که آیا روابط مشاهدهشده نسبت به تغییرات ذاتی دادهها مقاوم هستند یا خیر.
۷.۳ احتمالات پسین (Posterior Probabilities)
-
در تحلیل فیلوژنتیک بیزی، احتمالات پسین معیاری آماری برای پشتیبانی کلادها هستند که مستقیماً از فرایند نمونهگیری MCMC به دست میآیند.
-
هر درخت نمونهبرداری شده به توزیع پسین کمک میکند و نسبت درختهایی که شامل یک کلاد خاص هستند، احتمال صحت آن کلاد با توجه به دادهها و مدل را نشان میدهد.
-
تفاوت با بوتاسترپ:
-
وابسته به مدل و توزیعهای پیشین است.
-
برای گرههای با پشتیبانی ضعیف محافظهکارتر است و برای روابط با پشتیبانی قوی، اطمینان بالاتری ارائه میدهد.
-
-
روشهای بیزی امکان برآورد همزمان توپولوژی درخت، طول شاخهها و پارامترهای تکاملی را فراهم میکنند و عدم قطعیت را در سطوح مختلف استنتاج ادغام میکنند.
۷.۴ ارزیابی طول شاخهها و مدلهای جایگزینی
-
طول شاخهها در درخت فیلوژنتیک نشاندهنده میزان تغییر تکاملی در هر خط است.
-
تخمین دقیق طول شاخهها به ویژه در Maximum Likelihood و Bayesian اهمیت دارد، زیرا بر محاسبات نرخ جایگزینی، برآورد زمان واگرایی و تفسیرهای تکاملی بعدی تأثیر میگذارد.
-
ارزیابی کفایت مدل جایگزینی نیز اهمیت دارد.
-
معیارهای انتخاب مدل مانند Akaike Information Criterion (AIC) یا Bayesian Information Criterion (BIC) به شناسایی مدل مناسب برای دادهها کمک میکنند.
-
مدل نامناسب میتواند منجر به طول شاخههای نادرست، روابط جعلی و استنتاجهای تکاملی غلط شود.
-
-
ابزارهایی مانند ModelTest یا گزینههای داخلی IQ-TREE و MEGA امکان انتخاب سیستماتیک مدل قبل از ساخت درخت را فراهم میکنند.
۷.۵ مقایسه درختها و روشهای اجماعی (Consensus Methods)
-
تحلیل فیلوژنتیک اغلب منجر به چندین درخت قابل قبول میشود، به ویژه هنگام استفاده از جستجوی ابتکاری یا نمونهبرداری بیزی.
-
مقایسه درختها و خلاصهسازی شباهتها مرحلهای ضروری در ارزیابی است.
-
روشهای اجماعی یک درخت خلاصه تولید میکنند که ویژگیهای مشترک بین مجموعهای از درختها را نشان میدهد:
-
Strict Consensus Tree: تنها کلادهایی که در تمام درختهای ورودی هستند را نگه میدارد.
-
Majority-Rule Consensus Tree: کلادهایی را نگه میدارد که در درصد مشخصی (معمولاً ≥۵۰٪) از درختها ظاهر میشوند.
-
-
این روشها به شناسایی روابط پایدار در مقابل مبهم، مناطق با عدم قطعیت توپولوژی و نمایش خلاصه توزیع پسین یا بازنمونههای بوتاسترپ کمک میکنند.
۷.۶ تصویرسازی و تفسیر
-
نمایش بصری درختهای فیلوژنتیک برای تفسیر نتایج و ارتباط یافتهها ضروری است.
-
نرمافزارها و ابزارهای آنلاین مانند iTOL، ETE Toolkit و MEGA امکان نمایش طول شاخهها، مقادیر پشتیبانی بوتاسترپ یا پسین و حاشیهنویسی کلادها را فراهم میکنند.
-
تصویرسازی صحیح به شناسایی شاخههای بلند، پلیتومیها (گرههایی با بیش از دو فرزند) و مناطق با پشتیبانی ضعیف کمک میکند و راهنمای تصمیمگیری برای تحلیل بیشتر یا بهبود دادهها است.
-
تصویرسازی با کیفیت بالا به ویژه در ژنتیک تطبیقی، مطالعات تکاملی و انتشارات علمی اهمیت دارد، جایی که وضوح و دقت ضروری است.
بخش ۸: کاربردهای درختهای فیلوژنتیک
درختهای فیلوژنتیک تنها ساختارهای نظری نیستند؛ بلکه به ابزارهای اساسی در زمینههای متنوعی از جمله زیستشناسی، پزشکی، اکولوژی و ژنومیک تبدیل شدهاند. با فراهم کردن چارچوبی برای درک روابط تکاملی، این درختها به پژوهشگران کمک میکنند تا به بینشهایی درباره واگرایی گونهها، ژنومیک عملکردی، انتقال بیماریها و استراتژیهای حفاظت از گونهها دست یابند. کاربردهای تحلیل فیلوژنتیک از مطالعات میکروتکاملی در جمعیتها تا بازسازیهای ماکروتکاملی در سراسر درخت زندگی گسترده است. این بخش حوزههای اصلی استفاده از درختهای فیلوژنتیک را بررسی میکند و اهمیت آنها را در تحقیقات پایه و کاربردی نشان میدهد.
۸.۱ زیستشناسی تکاملی و مطالعات گونهزایی (Speciation)
-
در زیستشناسی تکاملی، درختهای فیلوژنتیک برای بازسازی تاریخچه زندگی و استنتاج الگوهای گونهزایی و سازگاری استفاده میشوند.
-
با تحلیل ویژگیهای مورفولوژیکی یا مولکولی، پژوهشگران میتوانند گروههای مونو فیلتیک را شناسایی کنند، ترتیب وقایع واگرایی را تعیین کنند و تکامل ویژگیهای خاص را ردیابی کنند.
-
مثال: تحلیل فیلوژنتیک فنچهای داروین، زمان و نحوه تکامل شکل منقار را در پاسخ به فشارهای محیطی روشن کرده است.
-
در مطالعات فیلوژنی پریماتها، روابط بین شاخههای اصلی مشخص شده و الگوهای رادیاسیون تطبیقی و همگرایی آشکار شده است.
-
درختهای فیلوژنتیک همچنین به زیستشناسان اجازه میدهند تا فرضیات تکاملی را آزمایش کنند، مانند اینکه آیا ویژگیهای مشابه در گونههای مختلف از طریق همگرایی تکاملی یا جد مشترک به وجود آمدهاند.
۸.۲ ژنومیک تطبیقی (Comparative Genomics)
-
درختهای فیلوژنتیک چارچوبی برای ژنومیک تطبیقی فراهم میکنند و امکان شناسایی ژنهای محافظتشده، عناصر تنظیمی و ویژگیهای ساختاری در گونههای مختلف را میدهند.
-
با نقشهبرداری توالیهای ژنی روی درخت فیلوژنتیک، پژوهشگران میتوانند وضعیت اجدادی ژنها را استنتاج کرده و الگوهای تکثیر، از دست رفتن و واگرایی ژنها را ردیابی کنند.
-
این روش به ویژه برای شناسایی ژنهای تحت انتخاب پاککننده (Purifying) یا مثبت (Positive) و درک پویایی تکاملی خانوادههای ژنی ارزشمند است.
-
روشهای فیلوژنتیک تطبیقی همچنین مطالعات تکامل معماری ژنوم مانند حفظ سینتنی (Synteny) و جابهجاییهای کروموزومی را تسهیل میکنند و بینشهایی درباره تکامل عملکردی و ساختاری ژنوم ارائه میدهند.
۸.۳ اپیدمیولوژی مولکولی (Molecular Epidemiology)
-
در پزشکی و بهداشت عمومی، درختهای فیلوژنتیک ابزاری حیاتی برای ردیابی انتشار بیماریهای عفونی و درک تکامل پاتوژنها هستند.
-
اپیدمیولوژیستهای مولکولی از تحلیل فیلوژنتیک برای بازسازی تاریخچه انتقال ویروسها و باکتریها، شناسایی منابع شیوع و پایش مقاومت دارویی استفاده میکنند.
-
مثالها:
-
ردیابی منشاء و گسترش جهانی HIV، آنفلوآنزا و SARS-CoV-2 که اطلاعات مهمی برای مداخلات بهداشت عمومی فراهم کرده است.
-
-
با تحلیل تنوع توالی در ژنوم پاتوژنها، پژوهشگران میتوانند گسترش کلونال، رخدادهای بازترکیبی و نقاط تکاملی فعال را شناسایی کنند که استراتژیهای طراحی واکسن، نظارت و کنترل بیماریهای عفونی را هدایت میکند.
۸.۴ زیستشناسی حفاظتی و تنوع زیستی (Conservation Biology & Biodiversity)
-
درختهای فیلوژنتیک نقش حیاتی در زیستشناسی حفاظتی ایفا میکنند و به اولویتبندی گونهها و جمعیتها برای حفاظت بر اساس متمایز بودن تکاملی کمک میکنند.
-
مفهوم تنوع فیلوژنتیک (Phylogenetic Diversity) بر حفظ شاخههایی که تاریخچه تکاملی منحصر به فرد دارند تأکید میکند، نه فقط شمارش تعداد گونهها.
-
مثال: تلاشهای حفاظتی که گونههای تکاملی متمایز و در معرض خطر جهانی (EDGE) را هدف قرار میدهند، از اطلاعات فیلوژنتیک برای حفاظت از تنوع تاکسونومی و ژنتیکی استفاده میکنند.
-
درختهای فیلوژنتیک همچنین در مدیریت زیستگاه، برنامههای بازگردانی و شناسایی گونههای مخفی (Cryptic Species) اطلاعات مهمی ارائه میدهند و استراتژیهای حفظ تنوع زیستی در اکوسیستمهای تهدیدشده را تقویت میکنند.
۸.۵ اکولوژی و تجمع جامعه (Ecology & Community Assembly)
-
در تحقیقات اکولوژیکی، درختهای فیلوژنتیک برای مطالعه تجمع جوامع و تعاملات گونهها استفاده میشوند.
-
با تحلیل خویشاوندی گونههای همزیست، اکولوژیستها میتوانند نقش تاریخچه تکاملی در شکلگیری جوامع اکولوژیکی را استنتاج کنند.
-
مثال:
-
گونههای نزدیک به هم ممکن است به دلیل تقاضای مشابه منابع رقابت شدیدی داشته باشند.
-
گونههای دور از هم ممکن است از طریق تفاوتهای جایگاه اکولوژیکی (Niche Differentiation) همزیستی کنند.
-
-
اطلاعات فیلوژنتیک همچنین به درک تکامل ویژگیها و تنوع عملکردی کمک میکند و نشان میدهد چگونه فرآیندهای تکاملی بر عملکرد و مقاومت اکوسیستمها تأثیر میگذارند.
۸.۶ پزشکی و فارماکوژنومیکس (Medicine & Pharmacogenomics)
-
در تحقیقات بالینی، درختهای فیلوژنتیک از فارماکوژنومیکس و پزشکی شخصیشده پشتیبانی میکنند، با ردیابی تاریخچه تکاملی ژنهای دخیل در متابولیسم دارو و حساسیت به بیماریها.
-
مثال: تحلیل فیلوژنتیک ژنهای سیتوکروم P450 میتواند الگوهای تنوع که بر اثربخشی و سمیت دارو در جمعیتهای مختلف تأثیر میگذارند را آشکار کند.
-
درختها همچنین به شناسایی ژنهای ارتولوگ برای مطالعات مدلهای آزمایشگاهی کمک میکنند و امکان تحقیقات ترجمهای از مدلهای آزمایشگاهی به سلامت انسان را فراهم میکنند.
-
علاوه بر این، در ژنومیک سرطان، روشهای فیلوژنتیک برای استنتاج تکامل تومور، ردیابی جمعیتهای کلونال و پیشبینی مقاومت به درمان استفاده میشوند و اهمیت فیلوژنتیک در پزشکی دقیق (Precision Medicine) را نشان میدهند.