تفسیر درختهای فیلوژنتیک
۱. مقدمهای بر فیلوژنتیک (Introduction to Phylogenetics)
فیلوژنتیک مطالعه علمی روابط تکاملی بین موجودات زنده، ژنها یا جمعیتها است. در اصل، فیلوژنتیک به دنبال آن است که بفهمد گونهها چگونه در طول زمان از اجداد مشترک منشعب شدهاند و چگونه ویژگیها، رفتارها یا توالیهای ژنتیکی تکامل یافتهاند. با بازسازی این روابط، دانشمندان میتوانند موجودات را در چارچوبی قرار دهند که تاریخچه تکاملی آنها را بازتاب دهد و این امر بینشهایی درباره منشأ تنوع زیستی، سازگاری و حتی سلامت انسان ارائه میدهد.
تحلیل فیلوژنتیک امروزه به یکی از ابزارهای اصلی زیستشناسی مدرن تبدیل شده است، زیرا راهی ساختاریافته برای تفسیر دادههای پیچیده زیستی فراهم میکند و ارتباطاتی را آشکار میسازد که از مشاهدات سطحی به راحتی قابل درک نیستند.
ریشههای تاریخی فیلوژنتیک را میتوان در آثار چارلز داروین در قرن نوزدهم جستجو کرد. داروین در کتاب منشأ گونهها (On the Origin of Species, 1859) پیشنهاد کرد که تمام موجودات زنده دارای جد مشترک هستند و مفهوم «درخت زندگی» (Tree of Life) را برای تصویرسازی الگوهای انشعاب تکاملی معرفی کرد. این استعاره، پایهای برای نمایش نظاممند روابط تکاملی شد، که بعدها با ظهور کلادیستیک (Cladistics) و فیلوژنتیک مولکولی (Molecular Phylogenetics) رسمیت یافت.
در مطالعات اولیه فیلوژنتیک، پژوهشگران عمدتاً بر ویژگیهای مورفولوژیکی قابل مشاهده مانند ساختار استخوانبندی، الگوهای رشد و ویژگیهای آناتومیک تکیه میکردند. این ویژگیهای فنوتیپی (Phenotypic Traits) شواهد اولیهای برای گروهبندی موجودات در طبقهبندیهای سلسلهمراتبی فراهم میکردند، اما محدودیتهایی داشتند؛ زیرا تکامل همگرا (Convergent Evolution) میتواند موجب شود گونههای نامرتبط به طور مستقل ویژگیهای مشابهی را تکامل دهند.
انقلاب مولکولی در قرن بیستم نقطه عطفی در فیلوژنتیک بود. با کشف ساختار DNA و امکان توالییابی (Sequencing) مواد ژنتیکی، دانشمندان به منبع عظیمی از دادهها دست یافتند. دادههای مولکولی شامل توالیهای DNA، RNA و پروتئین امکان بازسازی تاریخچههای تکاملی با دقت و وضوح بسیار بالاتر را فراهم کردند. درختهای فیلوژنتیک مبتنی بر دادههای مولکولی توانستند روابطی را آشکار سازند که پیشتر به دلیل محدودیتهای تحلیلهای مبتنی بر مورفولوژی پنهان مانده بودند، بهویژه در ریزاندامگان (Microorganisms) یا گونههای مخفی (Cryptic Species) که فاقد ویژگیهای فیزیکی آشکار هستند. امروزه فیلوژنتیک طیف وسیعی از رشتهها، از اکولوژی و زیستشناسی حفاظتی تا اپیدمیولوژی و ژنومیک را در بر میگیرد.
درختهای فیلوژنتیک، ابزار اصلی تحلیلهای فیلوژنتیک، نمایشهای گرافیکی روابط تکاملی هستند. هر شاخه (Branch) و گره (Node) در یک درخت اطلاعات کلیدی درباره اجداد مشترک و رویدادهای واگرایی ارائه میدهد. با تفسیر این درختها، پژوهشگران میتوانند ترتیب انشعاب خطوط تکاملی، زمانبندی رویدادهای تکاملی و الگوهای تکامل ویژگیها را استنتاج کنند.
علاوه بر جنبههای نظری، درختهای فیلوژنتیک کاربردهای عملی بسیاری دارند. برای نمونه، ردیابی روابط تکاملی پاتوژنها به دانشمندان امکان میدهد تا گسترش بیماریها را پیشبینی کنند، واکسنهای مؤثر طراحی کنند و مکانیزمهای مقاومت دارویی را بشناسند. همچنین، تحلیلهای فیلوژنتیک استراتژیهای حفاظتی را هدایت میکنند از طریق شناسایی گونههای تکاملی متمایز که برای حفظ تنوع زیستی حیاتی هستند.
فیلوژنتیک همچنین نقش مهمی در روشن کردن تاکسونومی (Taxonomy) دارد، علمی که به نامگذاری و دستهبندی موجودات زنده میپردازد. سیستمهای سنتی طبقهبندی اغلب موجودات را بر اساس شباهتهای سطحی گروهبندی میکردند که گاهی روابط تکاملی واقعی را به اشتباه نشان میداد. با استفاده از چارچوبی مبتنی بر اجداد و انشعابها، روشهای فیلوژنتیک به دانشمندان اجازه میدهد تا طبقهبندی تاکسونومیک را به گونهای بازسازماندهی کنند که بازتاب دقیقتری از تاریخچه تکاملی باشد. این رویکرد منجر به شناسایی دودمانهای جدید، بازتعریف جنسها و خانوادهها و درک بهتر فرآیندهای تکاملی ایجادکننده تنوع شده است.
در پایان، باید توجه داشت که فیلوژنتیک یک علم ایستا نیست. با جمعآوری دادههای جدید و توسعه روشهای تحلیلی پیشرفتهتر، درک ما از روابط تکاملی همواره در حال تغییر است. پیشرفتها در تکنولوژیهای توالییابی، الگوریتمهای محاسباتی و مدلهای آماری موجب افزایش دقت و مقیاس تحلیلهای فیلوژنتیک شده و امکان ساخت درختهای ژنومی عظیم شامل صدها یا هزاران گونه را فراهم کرده است. این حوزه همچنان در حال پیچیدهتر شدن و رشد است و دادهها را از منابع مختلف (مورفولوژیکی، مولکولی، اکولوژیکی و حتی رفتاری) یکپارچه میکند.
گسترش مداوم دانش فیلوژنتیک نه تنها درک ما از دنیای طبیعی را غنیتر میسازد، بلکه علوم کاربردی بسیاری را نیز پشتیبانی میکند و نشان میدهد که روشهای فیلوژنتیک همچنان ابزاری قدرتمند و ضروری در زیستشناسی مدرن هستند.
۲. مبانی درختهای فیلوژنتیک (Basics of Phylogenetic Trees)
یک درخت فیلوژنتیک نموداری است که فرضیههایی درباره تاریخچه تکاملی یک گروه از موجودات زنده، ژنها یا سایر موجودیتهای زیستی را نمایش میدهد. این نمودار صرفاً یک جدول شباهتها نیست، بلکه نمایشی ساختارمند از «نسب همراه با تغییر» (Descent with Modification) است که نشان میدهد چگونه دودمانها در طول زمان از یکدیگر منشعب شدهاند.
درک ساختار و معنای درختهای فیلوژنتیک برای تفسیر نتایج تحلیلهای تکاملی ضروری است، زیرا هر جزء از درخت معنای زیستی خاصی را منتقل میکند. برای مبتدیان، یک درخت ممکن است فقط خطوط منشعب به نظر برسد، اما برای زیستشناسان، این نمودار اطلاعات عمیقی درباره نیای مشترک، خویشاوندی و فرآیندهای ایجادکننده تنوع زیستی دارد.
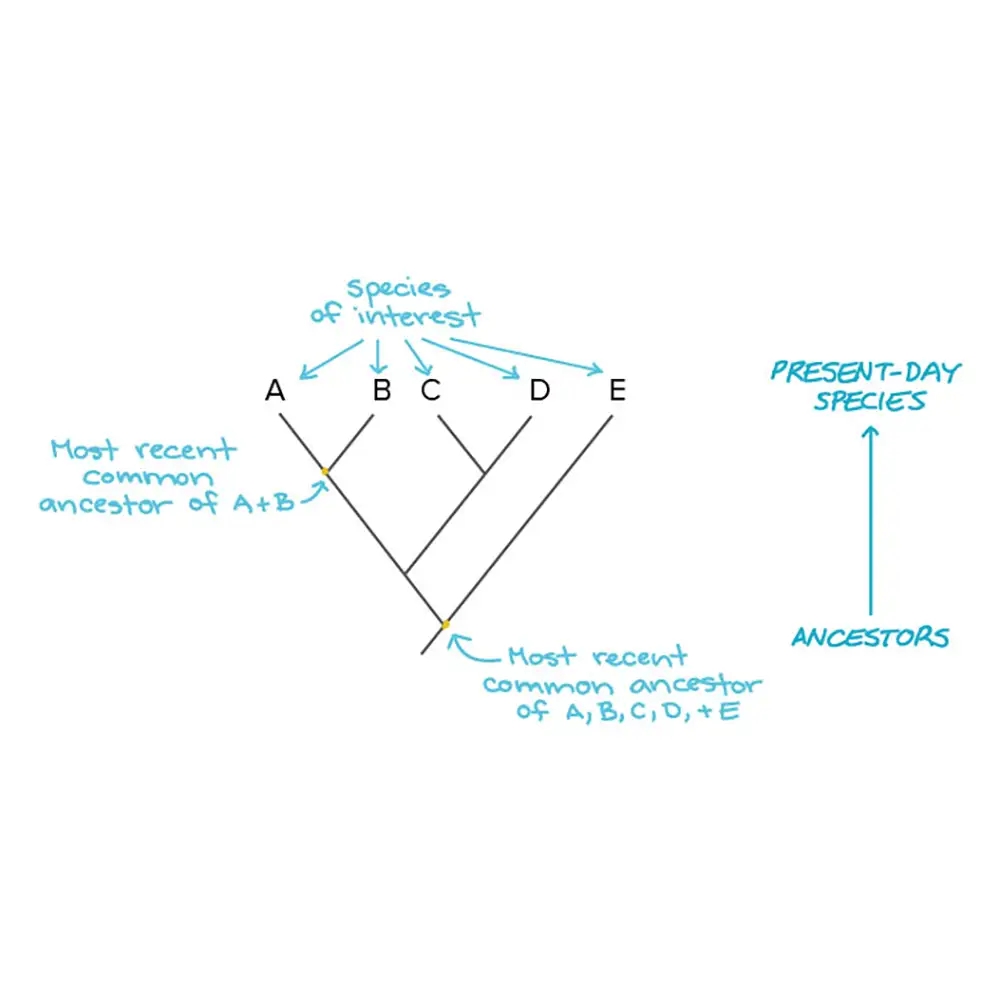
اجزای اصلی یک درخت فیلوژنتیک
در ابتداییترین سطح، یک درخت فیلوژنتیک شامل گرهها (Nodes)، شاخهها (Branches) و انتهاها (Tips یا Leaves) است:
-
گرهها (Nodes): نقاط واگرایی یا منشعب شدن هستند.
-
انتهاها (Tips/Terminal Nodes): نشاندهنده موجوداتی هستند که مطالعه میشوند، مثل گونههای زنده، فسیلها یا توالیهای ژنتیکی.
-
گرههای داخلی (Internal Nodes): نشاندهنده نیاکان مشترک فرضی هستند که از آنها انتهاها منشعب شدهاند. این اجداد معمولاً مستقیماً مشاهده نمیشوند بلکه بر اساس الگوهای شباهت و تفاوت میان انتهاها استنباط میشوند.
-
شاخهها (Branches): خطوطی که گرهها و انتهاها را به هم متصل میکنند و مسیر نسب و انشعاب را نشان میدهند. بسته به نوع درخت، شاخهها میتوانند زمان تکاملی، تغییر ژنتیکی، یا صرفاً ترتیب انشعابها را نمایش دهند.
ریشه و بیریشگی
-
ریشه (Root): نشاندهنده جد مشترک اخیر همه موجودیتهای درون درخت است. درختهای ریشهدار (Rooted Trees) دارای جهتمندی هستند و نشان میدهند چگونه دودمانها از یک جمعیت نیایی منشعب شدهاند.
-
درختهای بیریشه (Unrooted Trees): جد مشترک را مشخص نمیکنند و فقط روابط را بدون جهت تکاملی نمایش میدهند.
-
ریشهگذاری معمولاً نیازمند دادههای اضافی یا استفاده از «گروه بیرونی (Outgroup)» است. گروه بیرونی تاکسونی است که میدانیم نسبت به همه دیگر تاکسونها دورتر است و به عنوان مرجع برای تعیین ریشه استفاده میشود.
کلاد و گروههای تکاملی
یکی از مفاهیم کلیدی در تفسیر درختها کلاد (Clade) یا گروه تکتباری (Monophyletic Group) است. کلاد شامل یک نیای مشترک و همه نوادگان آن میشود.
-
تشخیص کلادها برای فهم روابط تکاملی بنیادی است، چون آنها گروههای طبیعی بر اساس نسب واقعی را نشان میدهند.
-
برای مثال، پستانداران یک کلاد تشکیل میدهند چون همه آنها از نیای مشترکی با ویژگیهایی مثل مو و غدد شیری منشعب شدهاند.
-
گروههایی که همه نوادگان نیای مشترک را شامل نمیشوند، پارافیلتیک (Paraphyletic) نامیده میشوند.
-
گروههایی که بر اساس ویژگیهای همگرا و نه نسب مشترک ساخته میشوند، پُلیفیلتیک (Polyphyletic) هستند.
تفسیر درست کلادها تضمین میکند که سیستمهای طبقهبندی بازتابدهنده واقعیت تکاملی باشند، نه شباهتهای سطحی.
انواع درختهای فیلوژنتیک
درختهای فیلوژنتیک انواع مختلفی دارند که هرکدام اطلاعات متفاوتی را نشان میدهند:
-
کلادوگرام (Cladogram): فقط ترتیب انشعاب دودمانها را نشان میدهد، بدون نمایش زمان یا میزان تغییر. همه شاخهها طول برابر دارند و تمرکز بر توپولوژی (Topology) است.
-
فیلوگرام (Phylogram): طول شاخهها متناسب با میزان تغییر تکاملی (مثل تعداد جانشینیها در توالی ژنتیکی) است.
-
کرونوگرام (Chronogram): طول شاخهها متناسب با زمان واقعی است، که معمولاً با شواهد فسیلی یا ساعت مولکولی (Molecular Clock) کالیبره میشود.
درک تفاوت این درختها حیاتی است، زیرا تفسیر نادرست طول شاخهها میتواند به نتیجهگیری غلط درباره روابط یا زمانبندی تکامل منجر شود.
توپولوژی و خواهر-تاکسونها
-
توپولوژی (Topology): الگوی انشعابها در درخت، مستقل از طول شاخهها.
-
توپولوژی تعیین میکند کدام تاکسونها به هم نزدیکترند.
-
اگر گونههای A و B قبل از اتصال به گونه C یک گره مشترک داشته باشند، A و B تاکسونهای خواهری (Sister Taxa) هستند.
توپولوژی حتی اگر طول شاخهها متفاوت باشد، همچنان روابط را به درستی نشان میدهد.
سبکهای نمایش درخت
درختهای فیلوژنتیک میتوانند به شکل افقی، عمودی یا دایرهای نمایش داده شوند. اما جهت نمایش هیچ تأثیری بر معنا ندارد.
-
چرخاندن شاخهها یا تغییر جهت درخت روابط را تغییر نمیدهد.
-
یکی از خطاهای رایج مبتدیان این است که فکر میکنند نزدیکی گونهها در چیدمان سمت چپ یا راست صفحه به معنای خویشاوندی بیشتر است. در واقع، فقط الگوی گرهها خویشاوندی را مشخص میکند.
اصطلاحات تکمیلی
-
پولیتومی (Polytomy): گرهای با بیش از دو شاخه فرعی.
-
اگر ناشی از عدم قطعیت دادهها باشد → پولیتومی نرم (Soft Polytomy).
-
اگر ناشی از انشعاب همزمان واقعی باشد → پولیتومی سخت (Hard Polytomy).
-
-
دودمانهای پایهای (Basal Lineages): شاخههایی که زودتر از بقیه از ریشه جدا شدهاند.
-
باید توجه داشت که اینها به معنای «ابتداییتر» یا «کمتر تکاملیافته» نیستند.
-
همه گونههای زنده به یک اندازه زمان تکاملی را از نیای مشترک طی کردهاند.
-
درختهای فیلوژنتیک = فرضیه
در نهایت باید دانست که درختهای فیلوژنتیک فرضیههایی درباره تاریخچه تکاملی هستند، نه حقیقت قطعی.
-
آنها از دادهها و مدلها ساخته میشوند که همیشه فرضیات و محدودیتهایی دارند.
-
با دادههای جدید یا روشهای بهتر، درختها ممکن است بازنگری شوند.
-
با این حال، وقتی درست خوانده شوند، چارچوبی قدرتمند برای درک الگوهای انشعاب حیات و مکانیزمهای ایجاد تنوع زیستی فراهم میکنند.
تسلط بر مبانی درختهای فیلوژنتیک، پایهای برای یادگیری عمیقتر درباره روشهای ساخت درخت، تفسیر طول شاخهها و کاربردهای گسترده فیلوژنتیک در زیستشناسی است.
۳. انواع درختهای فیلوژنتیک (Types of Phylogenetic Trees)
درختهای فیلوژنتیک میتوانند اشکال متفاوتی داشته باشند، بسته به اینکه چه نوع اطلاعاتی را قرار است منتقل کنند. هرچند همه درختها ساختار پایهای مشترکی از گرهها (Nodes)، شاخهها (Branches) و انتهاها (Tips) دارند، اما ظاهر و تفسیر آنها میتواند متفاوت باشد.
شناخت انواع درختهای فیلوژنتیک ضروری است زیرا نحوه رسم یک درخت تعیین میکند چه جنبههایی از تاریخچه تکاملی را میتوان استنباط کرد. برخی درختها بر ترتیب روابط تأکید دارند، در حالی که برخی دیگر بر میزان تغییرات یا گذر زمان تمرکز میکنند.
از یک مجموعه داده میتوان چندین نمایش مختلف ایجاد کرد، و توانایی تشخیص این اشکال تضمین میکند که نتیجهگیریها بر اساس معنای واقعی درخت باشد، نه بر اساس برداشت نادرست از ساختار آن.
۳.۱ درختهای ریشهدار و بیریشه (Rooted vs. Unrooted Trees)
یکی از بنیادیترین تمایزها بین درختها این است که ریشهدار باشند یا بیریشه:
-
درخت ریشهدار (Rooted Tree):
-
جد مشترک اخیر همه دودمانها را مشخص میکند.
-
جهتمندی به درخت میدهد و اجازه میدهد درباره ترتیب زمانی رویدادهای واگرایی نتیجهگیری کنیم.
-
مثال: در یک درخت ریشهدار از مهرهداران، ریشه نمایانگر جد مشترک همه مهرهداران است و شاخهها واگرایی آنها به ماهیها، دوزیستان، خزندگان، پرندگان و پستانداران را نشان میدهند.
-
بیشتر برای بازسازی تاریخچه تکاملی بر اساس نسب (Ancestry) استفاده میشوند.
-
-
درخت بیریشه (Unrooted Tree):
-
نیای مشترک را مشخص نمیکند.
-
فقط روابط دودمانها را بدون جهت تکاملی نشان میدهد.
-
در مراحل اولیه تحلیل تولید میشوند چون نیاز به فرضیات کمتری دارند.
-
برای تفسیر کامل باید با گروه بیرونی (Outgroup) یا شواهد خارجی ریشهگذاری شوند.
-
👉 هر دو شکل ارزشمندند، اما اهداف متفاوتی دارند و انتخاب بین آنها به ماهیت مطالعه بستگی دارد.
۳.۲ کلادوگرام، فیلوگرام و کرونوگرام
دستهبندی دیگر درختها بر اساس این است که آیا فقط الگوی انشعاب را نشان میدهند یا اطلاعات بیشتری مثل تغییر تکاملی یا زمان واقعی را نیز اضافه میکنند:
-
کلادوگرام (Cladogram):
-
سادهترین نوع درخت.
-
فقط ترتیب انشعاب دودمانها را نشان میدهد.
-
طول شاخهها اختیاری و بیمعنی است.
-
برای نمایش فرضیههای خویشاوندی مفید است، اما نمیتواند درباره سرعت تکامل پاسخ بدهد.
-
-
فیلوگرام (Phylogram):
-
طول شاخهها متناسب با میزان تغییر تکاملی (مثلاً تعداد جانشینیهای نوکلئوتیدی).
-
مفید برای مشاهده نرخ تغییرات و مقایسه میزان تکامل در دودمانهای مختلف.
-
-
کرونوگرام (Chronogram):
-
طول شاخهها متناسب با زمان واقعی است.
-
امکان تخمین زمان رویدادهای واگرایی را فراهم میکند.
-
نیاز به کالیبراسیون با شواهد فسیلی یا رویدادهای بیوژئوگرافی شناختهشده دارد.
-
👉 دانستن اینکه با کدام نوع درخت روبهرو هستیم حیاتی است، زیرا هرکدام استنباطهای متفاوتی از تاریخچه تکاملی ارائه میدهند.
۳.۳ درختهای توافقی و سوپرتریها
گاهی شواهد متناقض از دادهها یا روشهای مختلف به دست میآید. برای رفع این تناقضها از روشهای ترکیبی استفاده میشود:
-
درخت توافقی (Consensus Tree):
-
چندین درخت جایگزین را خلاصه میکند.
-
روابطی را که به طور مداوم حمایت میشوند حفظ کرده و روابط نامشخص را حلنشده باقی میگذارد.
-
مثال: اگر بیشتر تحلیلها نشان دهند گونههای A و B تاکسونهای خواهریاند، اما جایگاه گونه C مبهم باشد، درخت توافقی A و B را کنار هم قرار میدهد و C را بهطور نامطمئن نمایش میدهد.
-
یادآور این است که فرضیههای فیلوژنتیک قطعی نیستند، بلکه احتمالیاند.
-
-
سوپرتری (Supertree):
-
اطلاعات را از چندین درخت کوچکتر ترکیب میکند تا یک درخت جامعتر بسازد.
-
بهویژه در مقیاس بزرگ مثل ساخت «درخت زندگی (Tree of Life)» کاربرد دارد.
-
چون هیچ مجموعه دادهای شامل همه گونهها نیست، سوپرتریها به پژوهشگران اجازه میدهند تصویر کلیتری از تاریخچه تکاملی ترسیم کنند.
-
البته سوپرتری محدودیتها و خطاهای درختهای سازنده خود را نیز به ارث میبرد.
-
۳.۴ سبکهای بصری درختها
علاوه بر محتوا، سبک ترسیم درختها هم متفاوت است، هرچند اطلاعات بنیادی یکسان باقی میماند:
-
درختهای مستطیلی (Rectangular):
-
گرهها و شاخهها بهصورت جعبهای نمایش داده میشوند.
-
خوانایی بالایی دارند، بهویژه برای دادههای ساده.
-
-
درختهای قطری (Diagonal):
-
شاخهها مورب هستند.
-
برای دادههای بزرگتر جمعوجورترند.
-
-
درختهای شعاعی (Radial/Circular):
-
شاخهها به صورت دایرهای از مرکز منشعب میشوند.
-
برای نمایش مجموعههای داده بزرگ و پیچیده متعادلتر و زیباترند.
-
معمولاً در مطالعات ژنومی با صدها یا هزاران تاکسون استفاده میشوند.
-
👉 نوع نمایش بصری معنا را تغییر نمیدهد، اما میتواند الگوها را بسته به دادهها بهتر آشکار کند.
۳.۵ انواع تخصصی درختها
علاوه بر انواع استاندارد، شکلهای تخصصیتری هم وجود دارد که برای پرسشهای خاص تکاملی طراحی شدهاند:
-
درخت گونهها (Species Tree): روابط بین گونهها را نشان میدهد.
-
درخت ژنها (Gene Tree): تاریخچه تکاملی یک ژن خاص را نمایش میدهد.
-
به دلیل پدیدههایی مثل تکثیر ژن (Gene Duplication)، از دست رفتن ژن (Gene Loss) یا انتقال افقی ژن (Horizontal Gene Transfer)، درخت ژن همیشه با درخت گونهها تطابق ندارد.
-
درخت همتبارزایی (Coalescent Tree): در ژنتیک جمعیت برای ردیابی تبارها درون یک گونه تا رسیدن به جد مشترک اخیر استفاده میشود.
-
۴. روشهای ساخت درخت فیلوژنتیک (Methods of Phylogenetic Tree Construction)
ساخت درخت فیلوژنتیک صرفاً به معنای چیدن موجودات بر اساس شباهت ظاهریشان نیست. این کار یک فرآیند تحلیلی دقیق است که بر پایه دادههای زیستی، مدلهای ریاضی و استنباط آماری انجام میشود تا فرضیههایی درباره روابط تکاملی ارائه کند.
در طول یک قرن گذشته، روشهای متعددی برای بازسازی فیلوژنی توسعه یافتهاند که هرکدام نقاط قوت، محدودیتها و فرضیات خاص خودشان را دارند. برخی روشها بر اندازهگیری شباهت کلی تکیه میکنند، در حالی که برخی دیگر بر ویژگیهای مشتقشده مشترک (Shared Derived Traits) یا احتمال دادههای مشاهدهشده بر اساس یک مدل تکاملی تمرکز دارند.
👉 انتخاب روش مناسب به نوع دادهها، پرسشهای تکاملی و حتی منابع محاسباتی موجود بستگی دارد.
👉 درک اصول پشت این روشها برای تفسیر درختها و تشخیص شرایطی که نتایجشان قابل اعتمادتر است، ضروری است.
۴.۱ روشهای مبتنی بر فاصله (Distance-Based Methods)
یکی از قدیمیترین و شهودیترین رویکردها، روشهای مبتنی بر فاصله هستند که از اندازهگیری جفتی شباهت یا تفاوت بین تاکسونها برای ساخت درخت استفاده میکنند.
-
روش UPGMA (Unweighted Pair Group Method with Arithmetic Mean):
-
تاکسونها را مرحلهبهمرحله خوشهبندی میکند.
-
ابتدا نزدیکترین جفت (با کمترین فاصله ژنتیکی یا فنوتیپی) را ترکیب کرده و بهتدریج گروههای بزرگتر میسازد تا همه تاکسونها در یک درخت جای بگیرند.
-
فرض میکند که نرخ تکامل در همه دودمانها ثابت است (فرض ساعت مولکولی).
-
این روش ساده و سریع است، اما اگر نرخ تکامل بین دودمانها متفاوت باشد، میتواند نتایج گمراهکننده بدهد.
-
-
روش Neighbor-Joining (NJ):
-
مانند UPGMA از دادههای فاصلهای استفاده میکند، اما فرض نرخ تکامل ثابت را ندارد.
-
برای مجموعهدادههای بزرگ بسیار مؤثر است چون محاسباتی کارآمد دارد.
-
معمولاً درختهایی نزدیک به روابط واقعی تکاملی تولید میکند.
-
👉 مشکل اصلی روشهای فاصلهای این است که دادههای پیچیده را به یک آمار خلاصه تکبعدی تقلیل میدهند و ممکن است الگوهای مهم در تکامل ویژگیها (Character Evolution) نادیده گرفته شوند.
۴.۲ روشهای مبتنی بر ویژگی (Character-Based Methods)
برخلاف روشهای فاصلهای، این روشها از توزیع واقعی ویژگیها یا کاراکترها (مثلاً نوکلئوتیدها یا صفات ریختشناسی) بین تاکسونها استفاده میکنند.
-
بیشینه صرفهجویی (Maximum Parsimony, MP):
-
به دنبال درختی است که کمترین تعداد تغییر تکاملی را نیاز داشته باشد.
-
سادهترین فرضیهای را که با دادهها سازگار است انتخاب میکند (اصل صرفهجویی/Parsimony).
-
در توسعه کلادیستیک (Cladistics) نقش کلیدی داشت چون روشی نظاممند برای ساخت درخت از دادههای ریختشناسی ارائه داد.
-
محدودیت: به تکامل همگرا (Convergent Evolution) یا نرخهای نابرابر تغییر حساس است.
-
ممکن است دچار جذب شاخههای بلند (Long-Branch Attraction) شود، یعنی تاکسونهای دور به اشتباه نزدیک هم قرار بگیرند.
-
با وجود این محدودیتها، ابزار آموزشی مهمی است و گاهی وقتی دادهها کم یا روش بدون مدل نیاز باشد، استفاده میشود.
-
۴.۳ بیشینه درستنمایی (Maximum Likelihood, ML)
-
رویکردی آماری و دقیقتر نسبت به پارسیمونی است.
-
بر اساس احتمال مشاهده دادهها تحت یک مدل تکاملی مشخص عمل میکند.
-
بهجای کمینه کردن تغییرات، به عوامل زیر توجه میکند:
-
نرخهای متفاوت جانشینی نوکلئوتیدها،
-
تفاوت بین ترنزیشن (Transition) و ترنسورژن (Transversion)،
-
و حتی تغییرپذیری بین جایگاههای مختلف یک توالی.
-
-
این روش واقعگرایی زیستی بیشتری دارد و انعطافپذیرتر است.
-
محدودیت: بسیار محاسباتی سنگین است چون باید احتمال دادهها را برای درختهای متعدد محاسبه کند.
-
اما با رشد قدرت محاسباتی و الگوریتمهای کارآمد، امروز یکی از پرکاربردترین روشهاست.
👉 مزیت اصلی ML ترکیب واقعگرایی زیستی با دقت آماری است، بنابراین نتایج آن بسیار قابل اعتمادند.
۴.۴ استنباط بیزین (Bayesian Inference)
-
چارچوب درستنمایی را گسترش میدهد با افزودن احتمالات پیشین (Prior Probabilities).
-
بهجای یافتن یک درخت بهترین، توزیعی از درختهای ممکن تولید میکند که هرکدام با احتمال پسین (Posterior Probability) وزندهی شدهاند.
-
امکان میدهد که میزان اطمینان به هر کلاد محاسبه شود.
-
معمولاً از الگوریتمهای زنجیره مارکوف مونتکارلو (MCMC) برای کاوش فضای درختها استفاده میکند.
-
هزاران یا میلیونها درخت نمونهگیری میشوند و از آنها آمارهای خلاصه استخراج میشود.
-
مزیت: بهطور طبیعی اجازه میدهد اطلاعات پیشین مثل کالیبراسیونهای فسیلی یا روابط شناختهشده قبلی در تحلیل لحاظ شوند.
-
محدودیت: انتخاب نادرست پریورها یا مدلهای تکاملی میتواند نتایج را منحرف کند.
👉 با وجود این ملاحظات، استنباط بیزین بسیار محبوب شده است، بهویژه برای دادههای بزرگ و پیچیده که در آنها اندازهگیری عدمقطعیت به اندازه یافتن روابط مهم است.
۴.۵ دادههای ریختشناسی در برابر دادههای مولکولی
انتخاب نوع داده نیز بر روش ساخت درخت تأثیر میگذارد:
-
دادههای ریختشناسی (Morphological Data):
-
هم برای موجودات زنده و هم فسیلها کاربرد دارند.
-
امکان ادغام دودمانهای منقرضشده در بازسازی تکاملی را میدهند.
-
محدودیت: تحت تأثیر تکامل همگرا و دشواری تعریف کاراکترهای همولوگ بین گروههای بسیار متفاوت.
-
-
دادههای مولکولی (Molecular Data):
-
بسیار فراواناند و وضوح بالاتری در روابط ارائه میکنند، بهویژه در تاکسونهای نزدیک.
-
توالیهای DNA و پروتئین هزاران کاراکتر برای تحلیل آماری فراهم میکنند.
-
اغلب با مدلهای شناختهشده جانشینی سازگارند.
-
محدودیت: فسیلها داده مولکولی ندارند، پس تاریخ تکاملی عمیق همچنان به ریختشناسی یا ساعت مولکولی کالیبرهشده با فسیل متکی است.
-
👉 امروزه بسیاری از مطالعات رویکرد شواهد جامع (Total-Evidence Approach) را اتخاذ میکنند، یعنی ترکیب دادههای مولکولی و ریختشناسی برای حداکثر دقت.
۴.۶ نرمافزارها و پیادهسازی عملی
اجرای این روشها وابسته به ابزارهای نرمافزاری تخصصی است:
-
برای روشهای پارسیمونی و فاصلهای: MEGA، PAUP* و PHYLIP.
-
برای روشهای پیشرفته مثل ML و بیزین: RAxML، IQ-TREE، MrBayes و BEAST.
هرکدام نقاط قوتی دارند: از سرعت و مقیاسپذیری گرفته تا انعطاف در مدلسازی.
رابطهای کاربرپسند و منابع محاسباتی پرقدرت باعث شدهاند که تحلیل فیلوژنتیک دموکراتیک شود، یعنی پژوهشگران از رشتههای مختلف بتوانند بدون تخصص عمیق در زیستشناسی محاسباتی درخت بسازند.
👉 اما استفاده مسئولانه از این ابزارها نیازمند درک فرضیات زیرین است، چون نرمافزار خودکار نمیتواند جایگزین قضاوت علمی دقیق شود.
۵. تفسیر توپولوژی درخت
توپولوژی یک درخت تبارزایشی (Phylogenetic Tree) به ساختار شاخهبندی آن اشاره دارد که نشان میدهد تبارها (lineages) چگونه با یکدیگر مرتبط هستند. این جنبه بنیادیترین بخش یک درخت است، زیرا حتی بدون در نظر گرفتن طول شاخهها یا اطلاعات اضافه، خود توپولوژی به تنهایی فرضیههایی درباره نیاکان (ancestry) و تبار (descent) ارائه میدهد.
تفسیر درست توپولوژی نیازمند توجه به نحوهی قرارگیری گرهها (nodes)، شاخهها (branches) و تبارکها یا کلادها (clades) است و اغلب نتیجهگیریهای زیستی از درخت بر اساس همین ساختار صورت میگیرد.
تفسیر اشتباه توپولوژی میتواند منجر به فرضیات غلط درباره خویشاوندی، نیاکان یا فرایندهای تکاملی شود. بنابراین، داشتن درک روشن از توپولوژی برای دانشجویان و پژوهشگران حرفهای زیستشناسی تکاملی ضروری است.
گرهها (Nodes)
در مرکز توپولوژی، گرهها قرار دارند:
-
گرههای درونی (Internal nodes): نمایانگر نیاکان مشترک هستند.
-
گرههای انتهایی یا سرشاخهها (Terminal nodes / Tips): نمایانگر تاکسونها (taxa) یا گروههایی هستند که در تحلیل گنجانده شدهاند.
وقتی دو سرشاخه، یک گره درونی نزدیکتر را با هم به اشتراک بگذارند تا با تاکسونهای دیگر، آنها تاکسونهای خواهری (Sister taxa) محسوب میشوند.
این مفهوم کلیدی است زیرا نزدیکترین خویشاوندان را نشان میدهد.
مثلاً در یک درخت از نخستیها (Primates)، انسان و شامپانزه همواره بهعنوان تاکسونهای خواهری دیده میشوند که نشاندهندهی نیای مشترک اخیر آنهاست.
نکته مهم: تاکسونهای خواهری نسبی هستند و وابسته به دادههای موجود در درختاند. افزودن یا حذف تاکسونها میتواند روابط ظاهری را تغییر دهد؛ نه به این دلیل که تاریخچه واقعی عوض شده، بلکه چون چارچوب مقایسه تغییر کرده است.
کلاد یا گروههای تکتباری (Clades / Monophyletic groups)
کلاد (Clade) شامل یک نیاک مشترک و تمامی نوادگانش است. شناسایی کلادها برای تشخیص گروههای طبیعی و واقعی که بازتاب روابط تکاملی هستند ضروری است.
-
کلادها میتوانند بزرگ باشند (مثلاً شامل یک ردهی کامل از موجودات) یا کوچک (شامل یک جفت گونه خواهری).
-
کلادها زیربنای استفاده از درختهای تبارزایشی در ردهبندی (taxonomy)، سامانهشناسی (systematics) و زیستشناسی تطبیقی (comparative biology) هستند.
در مقابل:
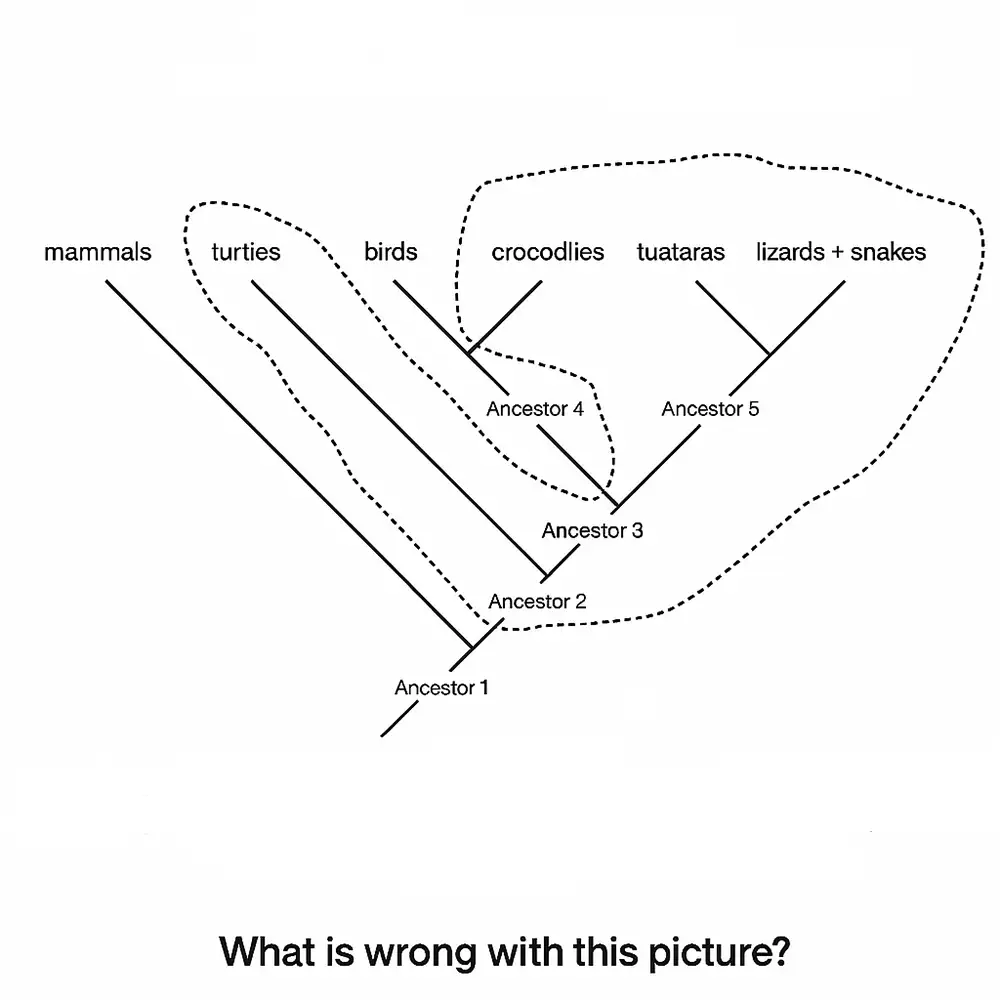
-
گروههای پَرتَباری (Paraphyletic): برخی نوادگان نیای مشترک را کنار میگذارند. (مثلاً خزندگان به شکل سنتی – بدون در نظر گرفتن پرندگان – یک گروه پرتَباری هستند.)
-
گروههای چندتباری (Polyphyletic): تبارهای نامرتبط را بر اساس ویژگیهای همگرایانه گرد هم میآورند.
تبارهای قاعدهای (Basal lineages)
تبارهای قاعدهای (Basal lineages) شاخههایی هستند که نزدیکتر به ریشهی درخت جدا شدهاند.
گاهی به اشتباه آنها را «ابتدایی» یا «کمتکاملیافته» توصیف میکنند، اما این برداشت غلط است.
همه گونههای زنده به یک اندازه زمان برای تکامل از نیای مشترکشان داشتهاند.
مثال: ماهیهای شُشدار (lungfish) بهعنوان یک تبار قاعدهای در مهرهداران شناخته میشوند، اما آنها موجوداتی بسیار تخصصیافته با تاریخ تکاملی طولانی هستند.
بنابراین، «قاعدهای» بودن به معنی «ابتدایی» بودن نیست.
چندشاخهایها (Polytomies)
گاهی درختها پُلیتومی (Polytomy) دارند، یعنی یک گره به سه یا چند شاخه تقسیم میشود.
-
پُلیتومی سخت (Hard polytomy): نشاندهندهی یک رویداد واقعی است که چندین تبار بهطور همزمان منشعب شدهاند (نادر، اما ممکن در مواردی مانند اشعاع انفجاری (rapid radiation)).
-
پُلیتومی نرم (Soft polytomy): بازتابدهندهی عدم قطعیت دادهها یا محدودیت روشها است. با دادههای بیشتر یا تحلیل بهتر، این چندشاخهایها میتوانند به شاخهبندیهای دوگانه (bifurcation) تفکیک شوند.
جهتگیری درخت (Tree orientation)
جهت رسم درخت اهمیتی ندارد.
درختها میتوانند چرخانده، قرینه یا برعکس شوند بدون اینکه معنای تکاملی آنها تغییر کند.
آنچه اهمیت دارد، ترتیب شاخهبندیهاست نه محل قرار گرفتن تاکسونها در سمت چپ یا راست.
اشتباه رایج: تصور اینکه تاکسونهای کنار هم در نمودار نزدیکتر هستند. در واقع، فقط گرهها و ارتباطاتشان نشاندهنده خویشاوندیاند.
درخت ژنی و درخت گونهای (Gene trees vs. Species trees)
-
درخت ژنی (Gene tree): تاریخچه یک ژن خاص را دنبال میکند.
-
درخت گونهای (Species tree): روابط بین گونهها را نشان میدهد.
به دلیل فرآیندهایی مثل چینش ناقص دودمانها (Incomplete lineage sorting)، تکثیر ژن (gene duplication) یا انتقال افقی ژن (horizontal gene transfer)، این دو همیشه منطبق نیستند.
مثلاً ممکن است درخت ژنی بعضی ژنهای سیستم ایمنی در نخستیها، انسان را با گوریل همگروه کند؛ در حالی که درخت گونهای نشان میدهد انسان به شامپانزه نزدیکتر است. این اختلاف بخشی از پیچیدگی تاریخ تکاملی است.
نسبت خویشاوندی (Relative vs. Absolute relatedness)
توپولوژی تنها نشان میدهد چه کسی با چه کسی نیای مشترک نزدیکتری دارد، نه اینکه چه کسی «بیشتر شبیه» دیگری است.
مثال: در یک درخت با انسان، شامپانزه و گوریل:
-
توپولوژی نشان میدهد انسان و شامپانزه نیاک مشترک نزدیکتری دارند.
-
اما این به معنای «شباهت بیشتر مطلق» یا «تکامل کمتر/بیشتر» نیست.
درختها بهعنوان فرضیه (Trees as hypotheses)
هر درخت یک فرضیه است، نه حقیقت مطلق.
درختهای مختلف میتوانند از دادهها یا روشهای متفاوت به دست آیند.
حتی توپولوژیهایی با پشتیبانی آماری بالا هم با شواهد جدید قابل بازنگریاند.
بنابراین، باید درختها را بهعنوان بهترین فرضیه موجود در نظر گرفت و آماده تغییر بود.
۶. طول شاخه و نرخهای تکاملی
در حالی که ساختار یا توپولوژی یک درخت فیلوژنتیک روابط بین گونهها را نشان میدهد، طول شاخهها اغلب اطلاعات اضافی درباره سرعت و میزان تکامل ارائه میکند. طول شاخهها صرفاً جنبه تزئینی یا دلخواه ندارند؛ بلکه جزئی جداییناپذیر در تفسیر بسیاری از درختهای فیلوژنتیک، بهویژه آنهایی که از دادههای مولکولی ساخته شدهاند، هستند. بسته به نوع درخت، طول شاخهها میتواند نشاندهنده تغییرات ژنتیکی، زمان واقعی یا هیچ معیار کمّی خاصی باشد. بنابراین درک صحیح طول شاخهها حیاتی است، زیرا برداشت نادرست میتواند به نتیجهگیریهای غلط درباره نرخهای تکاملی، زمانهای انشعاب یا میزان شباهت بین شاخهها منجر شود.
در سادهترین حالت، مانند کلا دوگرام (cladogram)، طول شاخهها هیچ معنایی ندارند. همه شاخهها با طول یکسان رسم میشوند و تنها هدف درخت، نمایش ترتیب شاخهبندی گونهها است. این نوع درخت روی توپولوژی تمرکز میکند و هیچ فرضی درباره میزان تغییر یا سرعت تکامل گونهها ندارد.
اما در فیلوگرامها (phylograms)، طول شاخهها متناسب با میزان تغییر تکاملی است، که معمولاً به صورت تعداد جایگزینیهای نوکلئوتیدی یا آمینواسیدی در دادههای مولکولی اندازهگیری میشود. بنابراین شاخه بلندتر نشاندهنده خطی است که تغییرات بیشتری از جد مشترک خود انباشته کرده است، و شاخه کوتاهتر نشاندهنده تغییرات کمتر است.
در کرونوگرامها (chronograms)، طول شاخهها متناسب با زمان مطلق است و تخمینی از زمان رخ دادن رویدادهای انشعاب ارائه میدهد. این تفاوتها نشان میدهد که تشخیص نوع درخت پیش از تفسیر طول شاخهها ضروری است.
ساعتهای مولکولی و تفسیر طول شاخهها
ساعت مولکولی یکی از مفاهیم کلیدی در تفسیر طول شاخهها در کرونوگرامها است. این فرضیه که در دهه ۱۹۶۰ توسط امیله زوکِرکاندل و لینوس پاولینگ پیشنهاد شد، بیان میکند که جهشهای ژنتیکی با نرخ نسبتاً ثابتی در طول زمان انباشته میشوند. اگر این فرض درست باشد، میزان اختلاف ژنتیکی بین دو شاخه میتواند برای تخمین زمان جد مشترک آنها استفاده شود.
ساعتهای مولکولی کاربردهای وسیعی دارند؛ از تخمین سن جد مشترک انسان و شامپانزه تا تاریخچه بزرگترین رویدادهای تکاملی مانند ظهور مهرهداران یا گیاهان گلدار.
با این حال، در عمل، نرخهای تکاملی معمولاً در همه شاخهها ثابت نیستند. برخی گروهها به دلیل زمان نسل کوتاهتر، نرخ جهش بالاتر یا فشارهای انتخابی شدید سریعتر تکامل مییابند، در حالی که گروههای دیگر کندتر تکامل میکنند. این تنوع نیازمند مدلهای پیشرفتهای است که امکان تغییر نرخ تکامل بین شاخهها را بدهند، نه اینکه صرفاً به ساعت مولکولی سخت تکیه کنند.
ساعت مولکولی آزاد (Relaxed Molecular Clock)
یکی از رویکردها برای مدیریت تغییر نرخها، استفاده از ساعت مولکولی آزاد است. به جای فرض یک نرخ ثابت برای تمام شاخهها، مدلهای ساعت آزاد به هر شاخه اجازه میدهند نرخ متفاوتی داشته باشد و اغلب این نرخها با توزیعهای احتمالاتی مدلسازی میشوند.
استنتاج بیزی (Bayesian inference) در توسعه این مدلها بسیار مؤثر بوده است، زیرا چارچوب آماری برای تخمین همزمان زمان انشعاب و تغییر نرخها فراهم میکند. ساعتهای آزاد توانایی ما در تاریخگذاری رویدادهای تکاملی را متحول کردهاند، اما نیاز به کالیبراسیون دقیق دارند تا نتایج قابل اعتماد ارائه کنند.
کالیبراسیون (Calibration)
کالیبراسیون فرآیند متصل کردن ساعت مولکولی به شواهد خارجی است تا تفاوتهای ژنتیکی نسبی به زمان مطلق ترجمه شوند. فسیلها رایجترین نقاط کالیبراسیون هستند، زیرا حداقل سن یک شاخه را براساس قدیمیترین نمونه شناختهشده ارائه میدهند.
مثلاً یک دایناسور پردار فسیلشده میتواند حداقل سن شاخه پرندگان را مشخص کند. رویدادهای زمینشناسی مانند شکلگیری پلهای زمینی یا جدایی قارهها نیز میتوانند نقاط کالیبراسیون باشند، اگر با زمان انشعاب شناختهشده همزمان باشند.
کالیبراسیون هم قدرتمند و هم چالشبرانگیز است:
-
نقاط کالیبراسیون کم → تخمینهای نادقیق
-
نقاط کالیبراسیون نامناسب → نتایج تحریفشده
بنابراین توجیه دقیق انتخاب نقاط کالیبراسیون به اندازه دادههای مولکولی اهمیت دارد.
شاخهها و تنوع نرخ تکامل
طول شاخهها میتواند نرخهای متفاوت تکامل را نشان دهد:
-
شاخههای بلند → تکامل سریع (به دلیل نرخ جهش بالا یا فشار انتخابی قوی)
-
شاخههای کوتاه → سکون تکاملی یا نرخ جهش پایین
مثلاً برخی ویروسها شاخههای بسیار بلندی دارند، زیرا نرخ جهش بالا و زمان نسل کوتاه دارند.
با این حال، تفسیر طول شاخهها نیاز به احتیاط دارد، زیرا عوامل دیگری مانند نمونهگیری ناقص، از دست رفتن ژنها یا انتقال افقی ژنها نیز میتوانند طول شاخهها را تحت تأثیر قرار دهند.
همچنین، شاخههای بلند میتوانند خطاهای ساخت درخت ایجاد کنند، بهویژه پدیده جذب شاخه بلند (long-branch attraction) که در آن شاخههای دور از هم با نرخ تغییر سریع به اشتباه کنار هم قرار میگیرند. شناخت این سوگیریها برای جلوگیری از برداشت غلط مهم است.
شاخهها و تفاوتهای انشعاب
شاخهها میتوانند نشاندهنده تفاوت در میزان تغییر بین شاخهها باشند:
-
اگر دو شاخه از جد مشترک جدا شده باشند ولی طول شاخهها متفاوت باشد، یکی از شاخهها تغییرات بیشتری نسبت به دیگری داشته است.
-
این تفاوت میتواند اطلاعاتی درباره تابع تطبیقی، رویدادهای دو برابر شدن ژنوم یا فشارهای اکولوژیکی متفاوت ارائه دهد.
مطالعات مقایسهای بین درختها با اطلاعات طول شاخه میتوانند توضیح دهند چرا برخی گروهها سریع تکثیر یافتهاند و برخی دیگر برای مدت طولانی تغییر کمی داشتهاند.
شاخههای داخلی کوتاه و انشعاب سریع
شاخههای داخلی کوتاه ممکن است نشاندهنده رویدادهای انشعاب سریع در تاریخ تکاملی باشند. وقتی چند شاخه از یک جد مشترک در مدت کوتاهی جدا شوند، شاخههای داخلی کوتاه نشاندهنده تغییر ژنتیکی کم قبل از انشعاب بعدی هستند.
این الگوها در رادیاسیونهای تطبیقی رایج است، جایی که فرصتهای اکولوژیکی باعث تنوع سریع شاخهها میشوند، مانند:
-
فنچهای داروینی در جزایر گالاپاگوس
-
ماهیهای سیکلید در دریاچههای آفریقایی
با این حال، شاخههای کوتاه داخلی چالشهایی برای بازسازی درخت ایجاد میکنند، زیرا تغییر ژنتیکی محدود باعث ابهام در ترتیب شاخهبندی میشود و ممکن است به چندشاخهای (polytomy) منجر شود.
محدودیتها و برداشتهای نادرست
مهم است بدانیم که تفسیر طول شاخهها همیشه به نوع درخت و فرضیات ساخت آن بستگی دارد:
-
یک فیلوگرام با فاصله ژنتیکی نمیتواند به عنوان کرونوگرام خوانده شود مگر اینکه به درستی کالیبره شده باشد.
-
تفاوت در طول شاخهها به معنای “تکامل بیشتر” نیست، زیرا همه شاخهها همچنان در حال تکامل هستند، هرچند با نرخها یا محدودیتهای متفاوت.
طول شاخهها اطلاعاتی درباره میزان تغییر یا گذر زمان میدهند، نه برتری، پیشرفت یا تکامل برتر.
آنچه درخت فیلوژنتیک نشان میدهد
درخت فیلوژنتیک یک فرضیه درباره «نزول با تغییر» (descent with modification) است: چگونگی تقسیم خطوط تکاملی از جد مشترک در طول زمان. هر گره داخلی نشاندهنده نقطهای در تاریخ است که در آن یک خط تکاملی به دو یا چند خط فرعی تقسیم شده است. شاخههای انتهایی (tips) نمایانگر نمونههای شما هستند: گونههای زنده، گونههای منقرضشده، جمعیتها یا توالیهای ژنی.
نکته مهم: درخت یک فرضیه علمی است و گرههای داخلی نشاندهنده نیستگان استنتاجی هستند، نه افراد واقعی، مگر در مواردی که فسیلها به صورت نمونه وارد شده باشند.
درخت ریشهدار vs. بدون ریشه
-
درخت ریشهدار: دارای ریشه مشخص است که جهت زمان را از ریشه به شاخهها نشان میدهد. تنها در این حالت میتوان گفت که «A و B جد مشترک نزدیکتری نسبت به A و C دارند».
-
درخت بدون ریشه: تنها روابط بین شاخهها را نشان میدهد، اما جهت تکاملی مشخص نیست. اغلب روشهای ساخت درخت ابتدا درختهای بدون ریشه تولید میکنند و سپس ریشهگذاری میشوند.
توپولوژی vs. هندسه
توپولوژی: ترتیب شاخهها و گرهها (چه کسی از چه کسی جدا شده است).
هندسه: نحوه رسم درخت (افقی، عمودی، دایرهای)؛ معمولاً معنای علمی ندارد مگر اینکه طول شاخهها معنادار باشند.
نکته: چرخاندن شاخهها در اطراف یک گره داخلی، توپولوژی را تغییر نمیدهد.
بخش دوم — گامبهگام خواندن درخت فیلوژنتیک
گام ۱ — تعیین اینکه شاخههای انتهایی نماینده چه چیزی هستند
-
آیا شاخهها نماینده گونهها، افراد، جمعیتها یا ژنها هستند؟
-
چرا مهم است: اشتباه گرفتن درخت ژنی با درخت گونهای میتواند به نتیجهگیریهای نادرست تکاملی منجر شود.
گام ۲ — بررسی ریشهدار بودن درخت
-
وجود ریشه: میتوانید جد مشترک اخیر هر مجموعه از شاخهها را مشخص کنید.
-
نبود ریشه: نمیتوانید جهت تکامل را مشخص کنید.
نکته عملی: اگر یک گونه به عنوان گروه بیرونی (outgroup) مشخص شده باشد، درخت ریشهدار است.
گام ۳ — درک دادهها و روش ساخت درخت
-
نوع داده: DNA، پروتئین، ویژگیهای مورفولوژیکی؟
-
ناحیه ژنی: mtDNA، rDNA، ژنوم کامل؟
-
روش استنتاج: فاصلهای (Neighbor-Joining)، بیشینه پارسیمونی (Maximum Parsimony)، بیشینه درستنمایی (Maximum Likelihood)، بیزین (Bayesian)؟
چرا مهم است: هر روش و داده نقاط قوت و محدودیت خاص خود را دارد.
گام ۴ — تعیین گروههای خواهری و کلادها
-
برای دو شاخه (A و B) مسیر هر شاخه را تا گره مشترک دنبال کنید؛ آن گره جد مشترک اخیر (MRCA) آنهاست.
-
اگر MRCA آنها جدیدتر از MRCA با شاخه دیگری (C) باشد، A و B با هم نزدیکترند.
نکته مهم: گروههای تکمنشا (monophyletic) همیشه شامل جد مشترک و همه فرزندان آن هستند.
گام ۵ — شناسایی گروههای تکمنشا (کلادها)
-
روش «برش تکتایی» (single-cut test): تصور کنید شاخهای را که کل گروه را به بقیه درخت متصل میکند قطع کنید. اگر با یک برش کلاد جد مشترک و همه فرزندان آن جدا شود، گروه تکمنشا است.
-
اگر نیاز به دو یا چند برش دارید، گروه غیرتکمنشا است (paraphyletic یا polyphyletic).
گام ۶ — بررسی طول شاخهها و مقیاس
-
اگر شاخهها متناسب با تغییر ژنتیکی یا زمان رسم شده باشند، طول شاخه معنادار است.
-
phylogram: طول شاخه = میزان تغییر ژنتیکی
-
chronogram: طول شاخه = زمان
-
cladogram: طول شاخه بیمعناست
نکته: بدون مقیاس، طول شاخهها را تفسیر نکنید.
گام ۷ — بررسی مقادیر اطمینان (support values)
-
Bootstrap: درصد حمایت گره در نمونهگیری مجدد. معمولاً >70–80% = حمایت متوسط، >90–95% = قوی.
-
Posterior probability (Bayesian): بین 0 و 1. مقدار بالای 0.95 نشاندهنده حمایت قوی است.
گام ۸ — در نظر گرفتن توپولوژیهای جایگزین
-
درخت ارائهشده یکی از فرضیههای ممکن است. ممکن است روشها یا دادههای دیگر نتایج متفاوت بدهند.
-
توصیه: اختلافها را بررسی کنید و به دلیل احتمالی آنها فکر کنید (مثلاً عدم تطابق ژنها، نمونهگیری ناقص، اشتباهات سیستماتیک).
گام ۹ — تفسیر بیولوژیکی
-
زیستجغرافیا: مهاجرت و تفکیک جغرافیایی را تحلیل کنید.
-
تکامل ویژگیها: حالتهای جدی ویژگیها و تکامل همگرا را شناسایی کنید.
-
زمانبندی: درختهای زمانی میتوانند سن جدها را تخمین بزنند.
-
حفاظت از گونهها: شناسایی خطوط تکاملی منحصربهفرد.
گام ۱۰ — ثبت محدودیتها و فرضیات جایگزین
-
هر نتیجه باید با اطمینان یا عدم اطمینان بیان شود.
-
محدودیتها: نمونهبرداری ناقص، طول توالی کوتاه، احتمال انتقال افقی ژنها، ترکیب گونهها.
بخش سوم — اشتباهات رایج
-
خواندن درخت از چپ به راست: اشتباه است، مرتبسازی شاخهها در صفحه معنای تکاملی ندارد.
-
اعتماد به طول شاخه بدون مقیاس: در cladogram طول شاخهها معنای علمی ندارند.
-
نادیده گرفتن مقادیر حمایت گرهها: نتیجهگیری بر گره ضعیف نادرست است.
-
تفسیر اشتباه درخت ژن به عنوان درخت گونهها: ممکن است به دلیل ILS، HGT یا هیبرید اشتباه شود.
-
اشتباه در درک گروههای غیرتکمنشا: Paraphyletic و Polyphyletic میتوانند در بیان روابط تکاملی گمراهکننده باشند.
بخش چهارشنبه— نکات پیشرفته
درخت ژن vs. درخت گونه
-
درخت ژن تاریخ یک ژن خاص را نشان میدهد.
-
درخت گونه تاریخ جدایی گونهها را نشان میدهد.
-
ممکن است اختلاف داشته باشند به دلیل: ILS، تکثیر/از دست رفتن ژن، HGT، هیبرید.
انتقال افقی ژن (HGT) و تکامل شبکهای
-
در باکتریها و ویروسها ممکن است درخت به شکل شبکه باشد، نه صرفاً درخت.
جذب شاخه طولانی
-
شاخههای بلند ممکن است به صورت مصنوعی به هم نزدیک شوند.
-
راه حل: استفاده از مدلهای تکاملی که تغییر نرخ را در نظر میگیرند.
بخش پنجم — جمعبندی
خواندن درخت فیلوژنتیک ترکیبی از دانش فنی و استدلال منطقی درباره جد مشترک است. با دنبال کردن چکلیست گامبهگام، میتوان:
-
شاخهها و کلادها را شناسایی کرد
-
طول شاخهها و مقادیر اطمینان را تفسیر کرد
-
محدودیتها و جایگزینها را ثبت کرد
-
تفسیرهای بیولوژیکی قابل اعتماد ارائه داد