درخت تبارزایی
I. مقدمهای بر فیلوژنی (Phylogenetics)
مطالعهی روابط تکاملی میان جانداران، همواره یکی از محورهای اصلی زیستشناسی بوده است. در قلب این تلاش علمی، دانشی به نام فیلوژنی (Phylogenetics) قرار دارد؛ دانشی که به بازسازی الگوهای شاخهبندی در تکامل میپردازد و گونهها، جمعیتها یا ژنها را در چارچوبی تاریخی جای میدهد که توضیح میدهد چگونه آنها از طریق نیای مشترک به یکدیگر متصل هستند. واژهی فیلوژنی از دو کلمهی یونانی phylon (به معنای تبار، نژاد یا گونه) و genesis (به معنای خاستگاه یا زایش) مشتق شده است و بهطور بنیادی با ردیابی تاریخچهی دودمانی حیات سر و کار دارد. فیلوژنی ابزاری فراهم میکند تا تنوع موجودات زنده را نه بهعنوان یک مجموعهی پراکنده و بینظم از اشکال مختلف، بلکه بهعنوان محصول تبار با تغییر (Descent with Modification) درک کنیم؛ جایی که شباهتها و تفاوتهای میان گونهها را میتوان از مسیرهای تکاملی آنها توضیح داد.
از زمان چارلز داروین که برای نخستین بار تاریخچهی تکاملی را بهصورت یک «درخت حیات» (Tree of Life) در کتاب خاستگاه گونهها (1859) به تصویر کشید، استعارهی درخت در مرکز زیستشناسی قرار گرفت. داروین یک نمودار ساده از ساختار شاخهای ترسیم کرد؛ خطوطی که از یک تنهی مشترک منشعب میشدند و نشان میدادند گونهها چگونه از نیاکان مشترک جدا شدهاند. این تصویر ساده اما عمیق، جوهرهی فیلوژنی را نشان میدهد: گونهها موجوداتی ایزوله و جدا از هم نیستند، بلکه شاخههایی از تاریخ عظیم و بههمپیوستهی حیات هستند. در اصطلاحات مدرن، فیلوژنی ابزارهایی فراهم میکند تا این شاخهها بازسازی شوند، جایگاه دقیق موجودات مشخص شود و زمان و آهنگ تغییرات تکاملی برآورد گردد.
در طول یک قرن گذشته، حوزهی فیلوژنی دگرگونیهای بنیادینی را تجربه کرده است. در آغاز، اندیشههای فیلوژنتیکی بیشتر بر مقایسههای مورفولوژیک و آناتومیکی تکیه داشتند؛ جایی که دانشمندان موجودات را بر اساس صفات ظاهری قابل مشاهده گروهبندی میکردند. در حالی که مورفولوژی همچنان یک منبع مهم اطلاعاتی است، ظهور زیستمولکولی و ژنومیک انقلابی در فیلوژنی ایجاد کرد. تحلیل توالیهای DNA، RNA و پروتئینها اکنون به زیستشناسان امکان میدهد روابط تکاملی را در بنیادیترین سطح سازمان زیستی بررسی کنند. این انقلاب مولکولی، بینشهایی را ارائه داد که با مورفولوژی به تنهایی دستیافتنی نبودند و اغلب موجب بازنگری در طبقهبندیهای سنتی و آشکار شدن روابط غیرمنتظره میان موجودات شدند. برای مثال، فیلوژنی مولکولی (Molecular Phylogenetics) نشان داد که نهنگها با سمداران جفتانگشتی (Artiodactyla) خویشاوندی نزدیکی دارند و پرندگان در واقع درون دودمان دایناسورها قرار دارند.
در مرکز این علم، درخت فیلوژنتیک (Phylogenetic Tree) قرار دارد؛ یک نمایش گرافیکی از این روابط. این نمودار، فرضیههایی دربارهی نیای مشترک و دودمانها را نشان میدهد، جایی که نقاط انشعاب یا گرهها (Nodes) بیانگر نیاکان مشترکاند و خطوط متصلکننده یا شاخهها (Branches) مسیرهای تکاملی موجودات را نمایان میسازند. درختها میتوانند در سطوح مختلف ساخته شوند: بین ژنها در یک ژنوم، بین افراد در یک جمعیت، بین گونهها در یک جنس، یا حتی میان گروههای ردهبندی بالاتر. مقیاس یک درخت فیلوژنتیک میتواند بسیار گسترده باشد (شامل همهی حیات روی زمین) یا بسیار محدود (فقط یک گروه کوچک از موجودات نزدیکخویشاوند). با این حال، همهی درختهای فیلوژنتیک هدف مشترکی دارند: نمایش ساختار تاریخ تکاملی.
تحلیل فیلوژنتیک تنها به مرتب کردن موجودات در یک نمودار شاخهای بسنده نمیکند. این تحلیل اندازهگیریهای کمی از فاصلهی تکاملی ارائه میدهد، ترتیب رویدادهای انشعاب را نشان میدهد و زمانهای واگرایی را برآورد میکند. با کالیبره کردن درختها با دادههای فسیلی یا ساعتهای مولکولی (Molecular Clocks)، دانشمندان میتوانند الگوهای شاخهبندی را به زمان زمینشناسی مطلق مرتبط کنند و تاریخنگاری حیات روی زمین را بازسازی نمایند. به این ترتیب، فیلوژنی بهطور عمیقی با دیرینشناسی، زمینشناسی و تکامل مولکولی پیوند خورده است و چارچوبی یکپارچه برای درک چگونگی پیدایش و تغییر تنوع زیستی در طول میلیاردها سال فراهم میکند.
اهمیت درختهای فیلوژنتیک فراتر از زیستشناسی تکاملی است. در پزشکی، از آنها برای ردیابی منشأ و گسترش عوامل بیماریزا استفاده میشود؛ برای مثال، در مطالعهی تکامل HIV یا ظهور سویههای جدید آنفلوانزا و ویروسهای کرونا. در زیستشناسی حفاظت (Conservation Biology)، معیارهای تنوع فیلوژنتیک کمک میکنند تا گونهها یا اکوسیستمها برای حفاظت اولویتبندی شوند؛ با این فرض که حفظ تاریخ تکاملی بیشتر، برای پایداری و گزینههای آینده ارزشمند است. در انسانشناسی، روشهای فیلوژنتیکی نهتنها برای بررسی تکامل انسان بلکه برای مطالعهی زبانها و فرهنگها نیز بهکار گرفته شدهاند؛ جایی که الگوهای شاخهبندیِ دودمانی بازتابی از تنوعیافتگی جوامع انسانی هستند. در زیستفناوری نیز درختهای تکاملی راهنمای کشف آنزیمهای جدید، آنتیبیوتیکها و مسیرهای ژنتیکی هستند، زیرا میتوانند به یافتن خویشاوندان سیستمهای زیستی شناختهشده کمک کنند.
به دلیل همین جایگاه محوری، درخت فیلوژنتیک به یکی از شناختهشدهترین نمودارها در علم بدل شده است. این نمودار صرفاً یک نمایش شماتیک نیست، بلکه یک فرضیهی تاریخی است؛ یک بیان علمی که میتواند آزموده، بازنگری و اصلاح شود، هنگامی که دادههای جدید در دسترس قرار گیرند. هر درخت فیلوژنتیک با نااطمینانی همراه است و روشهایی برای ارزیابی قابلیت اعتماد شاخههای آن توسعه یافتهاند؛ از جمله بوتاسترپینگ (Bootstrapping) و احتمالات پسین بیزی (Bayesian Posterior Probabilities). این سختگیری آماری تضمین میکند که تفسیرهای استخراجشده از درختها صرفاً گمانهزنی نباشند، بلکه استنتاجهای علمی معتبر و مبتنی بر داده باشند.
بنابراین، مطالعهی درختهای فیلوژنتیک ترکیبی از هنر و علم است. این مطالعه نیازمند ادغام دقیق نظریه، محاسبات و دادهها است، و همچنین مستلزم درکی عمیق از پیچیدگی فرآیندهای تکاملی. درخت هم یک استعاره و هم یک مدل است—راهی برای سازماندهی دانش زیستی و چارچوبی برای تولید فرضیات جدید. از این رو، فیلوژنی بخش جداییناپذیر زیستشناسی مدرن است و یکی از نیرومندترین ابزارها برای درک تنوع حیات به شمار میآید.
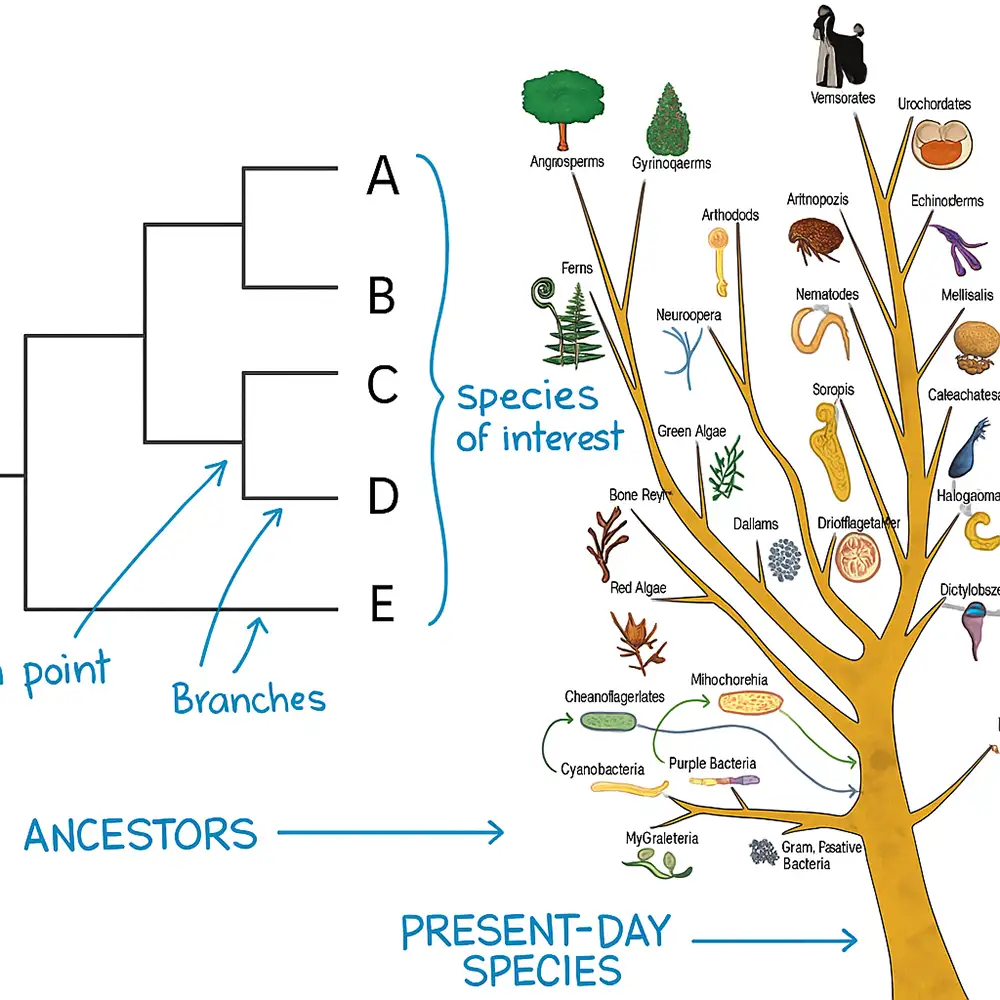
II. مفهوم درخت فیلوژنتیک
درخت فیلوژنتیک ابزار اصلی زیستشناسی تکاملی است و بهعنوان چارچوبی بصری و مفهومی برای فهم چگونگی ارتباط موجودات زنده از طریق نیاکان مشترک عمل میکند. در سادهترین سطح، درخت فیلوژنتیک یک نمودار است که فرضیات مربوط به روابط تکاملی بین مجموعهای از تاکسونها (که میتوانند گونهها، جمعیتها، افراد یا حتی ژنها باشند) را نشان میدهد. اگرچه استعاره «درخت» تداعی ساختار گیاهی میکند، اما در کاربرد علمی به یک نمودار شاخهای اشاره دارد که دگرگونی و انشعاب را در طول تکامل نشان میدهد؛ جایی که خطوط به سمت شاخههای مختلف انشعاب میکنند و گرهها (Nodes) نقاطی هستند که در آنها خطوط اجدادی منجر به پیدایش فرزندان میشوند. درختهای فیلوژنتیک تصاویر مستقیم تاریخ نیستند؛ یعنی شبیه یک عکس یا ثبت فسیلی عمل نمیکنند، بلکه استنتاجهایی هستند که از دادههای موجود استخراج شده و به شکل یک فرضیه منسجم درباره چگونگی تنوع حیات در طول زمان ارائه میشوند.
مفهوم درخت فیلوژنتیک بر اساس ایدهی اصلی داروین درباره «درخت زندگی» ساخته شده، اما درختهای مدرن جزئیات و دقت بسیار بیشتری دارند. این درختها نه تنها ترتیب انشعاب خطوط تبار را نشان میدهند، بلکه در بسیاری از موارد زمان نسبی یا مطلق رویدادهای واگرایی، میزان تغییرات ژنتیکی و ساختار کلادها را نیز شامل میشوند. از این نظر، درخت فیلوژنتیک هم نقشهای از روابط و هم یک مدل کمی از تکامل است. درخت اطلاعاتی درباره نیاکان – چه کسی با چه کسی مرتبط است – و فرآیند – سرعت واگرایی خطوط تبار، زمان وقوع نوآوریهای تکاملی مهم و ارتباط بین گروهها ارائه میدهد.
یکی از نکات مفهومی بسیار مهم این است که درخت فیلوژنتیک یک فرضیه است، نه حقیقت قطعی. دادههای مختلف یا روشهای تحلیلی متفاوت ممکن است درختهای متفاوتی ارائه دهند و زیستشناسان اغلب چندین درخت را مقایسه میکنند تا استحکام نتایج خود را ارزیابی کنند. برای مثال، دادههای مولکولی ممکن است یک ترتیب روابط میان گروهی از گونهها را نشان دهند، در حالی که ویژگیهای مورفولوژیکی ترتیب دیگری را پیشنهاد کنند. حل این اختلافات نیازمند بررسی دقیق شواهد است و معمولاً منجر به مدلهای دقیقتر و پیچیدهتر میشود. بنابراین، درخت فیلوژنتیک بهترین شکل بهعنوان چارچوبی قابل آزمون است که با کشف دادههای جدید میتواند بازبینی شود.
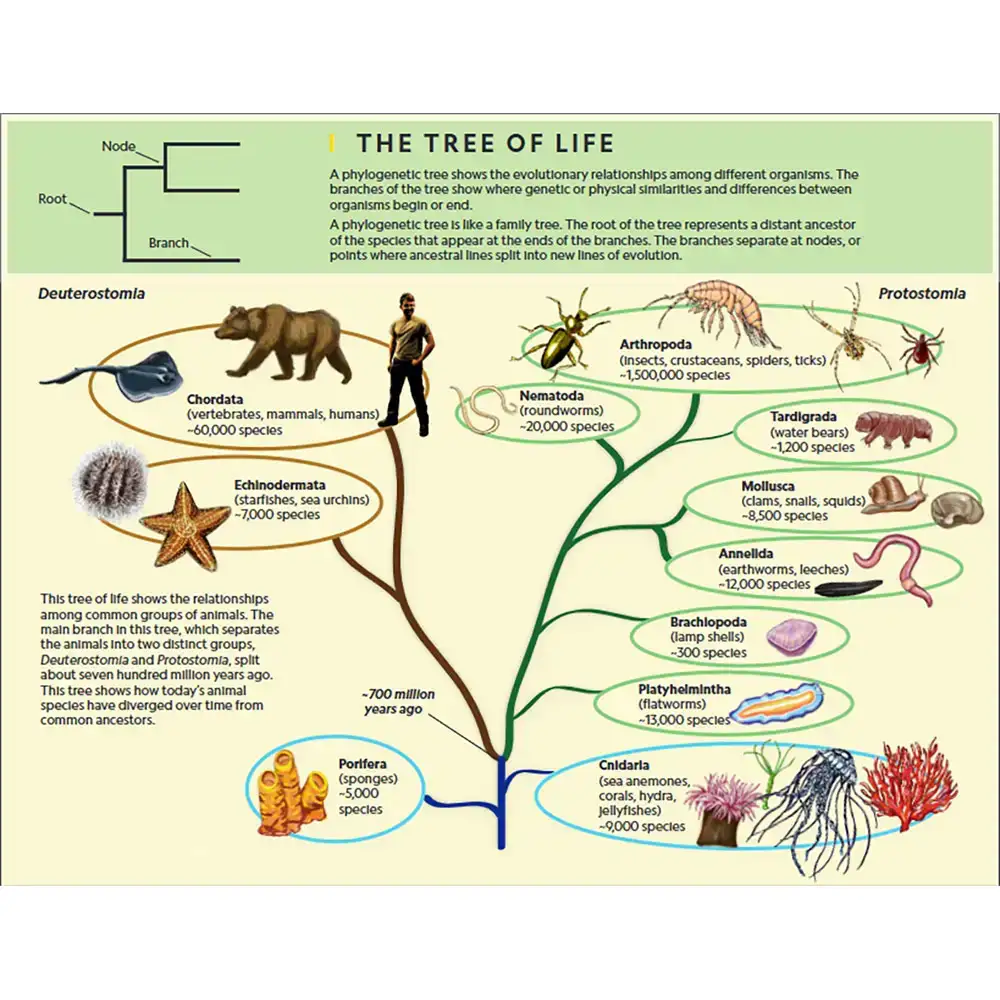
یک درخت از چند مولفه اصلی تشکیل شده است که به آن معنا میدهند. نوکها (Tips یا Leaves) نمایانگر تاکسونهای مورد مطالعه هستند – گونهها، افراد یا توالیها. گرههای داخلی (Internal Nodes) نیاکان مشترک را نشان میدهند که معمولاً فرضی و مستقیماً مشاهده نشدهاند اما از دادهها استنتاج میشوند. شاخهها (Branches) این گرهها را به هم متصل میکنند و خطوط تبار را نشان میدهند و گاهی طول آنها با میزان تغییر ژنتیکی یا زمان مقیاسبندی میشود. در پایه درخت، ریشه (Root) قرار دارد که نمودار را تثبیت میکند و آخرین نیاکان مشترک همه تاکسونها را مشخص میکند. درختها میتوانند ریشهدار (Rooted) باشند و جهتگیری از نیاکان به فرزندان را نشان دهند، یا بیریشه (Unrooted) باشند و فقط روابط را بدون مشخص کردن نیاکان نمایش دهند. چیدمان شاخهها یا توپولوژی (Topology) ویژگی مرکزی است که فرضیات تکاملی را منتقل میکند.
باید توجه داشت که درختهای فیلوژنتیک پیشرفت یا روند خطی را نشان نمیدهند. یک تصور نادرست رایج این است که درخت را بهصورت نردبان بخوانیم، با موجودات «ابتدایی» در پایین و موجودات «پیشرفته» در بالا. این تفسیر گمراهکننده و از نظر علمی نادرست است. تمام نوکهای درخت به یک اندازه تکامل یافتهاند، به این معنا که هر خط تبار تاریخچه تغییرات خود را از زمان واگرایی از نیاکان مشترک طی کرده است. برای مثال، انسانها «بیشتر تکامل یافته» از قورباغهها نیستند؛ هر دو یک نیاکان مشترک دارند و مسیرهای تکاملی متفاوتی را دنبال کردهاند. بنابراین، درخت باید بهعنوان الگوی روابط، نه رتبهبندی برتری، خوانده شود.
یک ویژگی مفهومی دیگر تفاوت بین گروههای مونوفیلتیک (Monophyletic)، پارافیلتیک (Paraphyletic) و پلیفیلتیک (Polyphyletic) است. گروه مونوفیلتیک یا کلاد (Clade) شامل یک نیاکان و همه فرزندان آن است و واحد ترجیحی در سیستماتیک مدرن محسوب میشود، زیرا تاریخچه واقعی تکاملی را منعکس میکند. گروه پارافیلتیک شامل نیاکان و بخشی از فرزندان است و گروه پلیفیلتیک از تاکسونهایی تشکیل شده که نیاکان مشترک نزدیکی در داخل گروه ندارند. برای مثال، خزندگان (Reptiles) بهطور سنتی یک گروه طبیعی در نظر گرفته میشدند، اما وقتی پرندگان (Birds) حذف شوند، «خزندگان» پارافیلتیک میشوند، زیرا پرندگان یک نیاکان مشترک با آنها دارند. شناخت این تفاوتها برای تفسیر صحیح درختهای فیلوژنتیک ضروری است.
درختها میتوانند انواع و نمایشهای متفاوتی داشته باشند. کلادوگرام (Cladogram) فقط ترتیب انشعابها را نشان میدهد و به میزان تغییرات تکاملی یا زمان توجهی ندارد. فیلوگرام (Phylogram) طول شاخهها را متناسب با میزان تغییر ژنتیکی نشان میدهد و حس سرعت تکامل در خطوط مختلف را ارائه میدهد. درخت اولترامتر (Ultrametric Tree) زمان را بهطور صریح نشان میدهد و همه نوکها به زمان حال تراز میشوند، بنابراین امکان استنتاج زمان واگرایی وجود دارد، اگر از ساعت مولکولی (Molecular Clock) یا کالیبراسیون فسیلی استفاده شود. هر نمایش برای هدف متفاوتی استفاده میشود و انتخاب آن بستگی به نوع داده و پرسش تحقیق دارد.
جهت درخت نیز انعطافپذیر است. درختها میتوانند عمودی، افقی یا حتی شعاعی ترسیم شوند، اما جهت تاثیری بر معنا ندارد. آنچه اهمیت دارد ترتیب انشعابها است، نه اینکه یک تاکسون در سمت چپ یا راست نمودار قرار دارد. برای مثال، چرخاندن افقی درخت تفسیر آن را تغییر نمیدهد، هرچند ممکن است روی سهولت دیداری اثر بگذارد. این نکته نشان میدهد که درختهای فیلوژنتیک اشیاء ریاضی هستند که میتوان آنها را به اشکال بصری مختلف نمایش داد.
یکی از جنبههای قدرتمند درختهای فیلوژنتیک این است که میتوانند در مقیاسهای مختلف زیستشناسی مقیاسبندی و تو در تو شوند. در سطح ژن، یک درخت ممکن است نشان دهد چگونه نسخههای مختلف یک خانواده ژنی از طریق رویدادهای تکثیر پدید آمدهاند. در سطح جمعیت، درخت میتواند روابط شجرهای بین افراد را با استفاده از دادههایی مانند DNA میتوکندریایی یا میکروساتلایتها نمایش دهد. در سطح گونهها، درختها تنوع خطوط تبار از طریق گونهزایی را نشان میدهند. در سطوح بالاتر، میتوانند الگوهای گسترده تاریخچه حیات، مانند واگرایی پستانداران، پرندگان و خزندگان، را نشان دهند. عمومیت ساختار درخت آن را برای کاربرد در تمام مقیاسها مناسب میکند، از کوچکترین واحدهای ژنتیکی تا گستردهترین چشمانداز درخت زندگی.
تفسیر یک درخت فیلوژنتیک نیازمند توجه به الگوهای انشعاب در مقابل طول شاخهها است. توپولوژی روابط را نشان میدهد، اما طول شاخهها ممکن است اطلاعات اضافی درباره میزان تغییر یا زمان را ارائه کنند. در برخی درختها، طول شاخهها دلخواه هستند و نباید بیش از حد تفسیر شوند؛ در دیگر درختها، اطلاعات مهمی درباره نرخهای تکامل یا زمانهای واگرایی فراهم میکنند. برای مثال، در فیلوگرام ساختهشده از توالیهای DNA، شاخه طولانیتر معمولاً نشاندهنده واگرایی ژنتیکی بیشتر است. در مقابل، در کلادوگرام، تمام شاخهها ممکن است یکسان کشیده شوند و فقط ترتیب تقسیمها را تأکید کنند. درک نوع درخت برای جلوگیری از برداشت نادرست ضروری است.
ریشهگذاری (Rooting) درخت اهمیت ویژهای دارد، زیرا جهت تاریخچه تکاملی را مشخص میکند. بدون ریشه، درخت بیریشه است و تنها روابط تاکسونها را نشان میدهد و مشخص نمیکند کدام گرهها نیاکان هستند. ریشهگذاری معمولاً نیازمند اضافه کردن یک اوتگروپ (Outgroup) است؛ تاکسونی که خارج از گروه مورد نظر اما هنوز مرتبط با آن است. با مقایسه تاکسونهای گروه مورد مطالعه (Ingroup) با اوتگروپ، میتوان جایگاه ریشه را تعیین کرد. این مرحله حیاتی است، زیرا ترتیب استنتاجی رویدادهای واگرایی را مشخص میکند. برای مثال، ریشهگذاری یک درخت مهرهداران با لنسلت (Lancelet)، یک کوردات پایهای (Basal Chordate)، به زیستشناسان امکان میدهد بفهمند ماهیهای بدون فک قبل از پیدایش مهرهداران دارای فک واگرا شدهاند.
کاربردهای درختهای فیلوژنتیک فراتر از نمودارهای ساده اجداد و فرزندان است. آنها بهعنوان ابزارهایی برای آزمون فرضیات تکاملی عمل میکنند. برای مثال، توزیع ویژگیها روی درخت میتواند بررسی شود تا مشخص شود آیا ویژگیهای مشابه مستقل ایجاد شدهاند یا از نیاکان مشترک به ارث رسیدهاند. این روش، معروف به نقشهگذاری ویژگیها (Character Mapping)، برای مطالعه تکامل ویژگیهای پیچیده مانند بالها، چشمها یا فتوسنتز استفاده شده است. همچنین، روشهای مقایسهای فیلوژنتیک (Phylogenetic Comparative Methods) از ساختار درخت استفاده میکنند تا همبستگی بین ویژگیها را با در نظر گرفتن نیاکان مشترک بررسی کنند و به دانشمندان امکان میدهد سؤالاتی درباره سازگاری، تنوع و جایگاههای اکولوژیکی را بررسی کنند.
در سطح فلسفیتر، مفهوم درخت فیلوژنتیک ماهیت سلسلهمراتبی تاریخ حیات را به تصویر میکشد. الگوی انشعاب نشان میدهد که تنوع زیستی مجموعهای از اشکال غیرمرتبط نیست، بلکه ساختاری به هم پیوسته است که از طریق واراثت شکل گرفته است. این دیدگاه کاملاً متفاوت از دیدگاههای پیشداروینی در طبقهبندی موجودات است که اغلب آنها را در زنجیرههای خطی یا دستهبندیهای ایستا قرار میدادند. درخت تأکید میکند که روابط نسبی هستند، نه مطلق، و هر گونه هم منحصربهفرد و هم مرتبط است. به این ترتیب، درخت فیلوژنتیک نه تنها یک مدل علمی بلکه یک چارچوب مفهومی است که نحوه تفکر ما درباره جهان زنده را شکل میدهد.
در پایان، مفهوم درخت فیلوژنتیک نظریه، داده و تصویرسازی را در یک مدل منسجم از روابط تکاملی ترکیب میکند. این درخت از عناصر اساسی – نوکها، گرهها، شاخهها، ریشهها، کلادها و توپولوژی – تشکیل شده است که با هم فرضیات اجداد و واراثت را نشان میدهند. درختها میتوانند اشکال مختلفی داشته باشند – کلادوگرام، فیلوگرام، درخت اولترامتر – اما همه برای سازماندهی تنوع زیستی در چارچوب تاریخی استفاده میشوند. با رد تصورات غلط درباره پیشرفت خطی و تأکید بر برابری تمام خطوط تبار، درخت فیلوژنتیک راهی منسجم و دقیق برای درک پیچیدگی حیات ارائه میدهد. این مفهوم یکی از قدرتمندترین و پایدارترین مفاهیم زیستشناسی است؛ یک زبان نموداری که داستان تکامل را از طریق آن میتوان روایت و آزمایش کرد.
III. اجزای یک درخت تبارزایشی
ساختار یک درخت تبارزایشی ممکن است از نظر ظاهری بسیار ساده به نظر برسد و اغلب تنها شامل خطوط و نقاط انشعاب روی صفحه است. اما در زیر این سادگی بصری، مجموعهای غنی از مفاهیم و معانی روششناختی و مفهومی نهفته است. هر عنصر درخت نشاندهنده یک رابطه تکاملی یا استنتاج مشخص است. برای تفسیر صحیح یک درخت، لازم است اجزای بنیادی آن—گرهها، شاخهها، ریشهها، کلادها، اوتگروهها و توپولوژی—بهخوبی درک شوند. این عناصر با هم دستور زبان تبارزایشی را تشکیل میدهند، یعنی قوانینی که بر اساس آنها فرضیههای تکاملی بیان و درک میشوند.
۱. گرهها (Nodes)
گرهها نقاط تقاطع در یک درخت تبارزایشی هستند و نشاندهنده عناصر حیاتی در تاریخچه تکاملی شاخهها محسوب میشوند. آنها به دو نوع اصلی تقسیم میشوند: گرههای خارجی (External/Terminal Nodes) و گرههای داخلی (Internal Nodes).
-
گرههای خارجی (External Nodes): که به برگها یا نوکها نیز معروفاند، مربوط به تاکسونهای واقعی وارد شده در تحلیل هستند. این تاکسونها میتوانند شامل ارگانیسمهای فردی، جمعیتها، گونهها یا ژنها باشند، بسته به محدوده مطالعه.
-
مثال: در یک درخت تبارزایشی پستانداران، هر گره خارجی میتواند نشاندهنده یک گونه خاص مانند Homo sapiens، Pan troglodytes (شامپانزه) یا Canis lupus (گرگ) باشد.
-
در یک درخت ژنی، نوکها ممکن است نشاندهنده توالیهای مشخصی باشند که از گونهها یا افراد مختلف نمونهبرداری شدهاند.
-
نکته مهم: گرههای خارجی درخت را به واقعیت مشاهدهشده متصل میکنند، زیرا آنها مربوط به دادههای نمونهبرداریشده هستند و موجودات فرضی نیستند.
-
-
گرههای داخلی (Internal Nodes): نمایانگر نیاکان مشترک فرضی تاکسونهای مرتبط هستند. این گرهها بهطور مستقیم مشاهده نمیشوند بلکه از دادهها و روش ساخت درخت استنتاج میشوند.
-
مثال: در درختی شامل انسانها، شامپانزهها و گوریلها، گره داخلی که انسان و شامپانزه را به هم پیوند میدهد، آخرین نیای مشترک آنها را نشان میدهد که میلیونها سال پیش میزیسته است.
-
اهمیت: گرههای داخلی در تفسیر روابط تکاملی اهمیت محوری دارند، زیرا نقاط انشعابی که منجر به تنوع کنونی شدهاند را مشخص میکنند.
-
گرهها میتوانند اطلاعات اضافی نیز داشته باشند:
-
در درختهای کالیبرهشده بر اساس زمان، موقعیت گره داخلی ممکن است متناظر با زمان برآوردی انشعاب باشد که اغلب توسط فسیلها یا مدلهای ساعت مولکولی پشتیبانی میشود.
-
در درختهایی با مقیاس طول شاخهها، گرهها میتوانند تجمع تغییرات ژنتیکی را نشان دهند.
جمعبندی: در حالی که گرههای خارجی درخت را به واقعیت نمونهبرداریشده متصل میکنند، گرههای داخلی پنجرهای به گذشته عمیق ارائه میدهند و فرضیههایی درباره نیاکان ارائه میکنند که بهطور مستقیم قابل دسترسی نیستند.
۲. شاخهها (Branches / Edges)
شاخهها یا یالها، گرهها را به هم متصل میکنند و نمایانگر شاخههایی هستند که تغییرات تکاملی در آنها رخ میدهد. از نظر مفهومی، یک شاخه میتواند به عنوان مسیر تکاملی از یک نیای مشترک تا نوادگان آن در نظر گرفته شود و شامل تغییرات ژنتیکی و فنوتیپی است که در طول زمان انباشته شدهاند.
-
انواع شاخهها:
-
شاخههای داخلی: دو گره داخلی را به هم متصل میکنند و نمایانگر شاخههایی هستند که بین رویدادهای انشعاب متوالی وجود داشتهاند.
-
شاخههای ترمینال: یک گره داخلی را به یک گره خارجی متصل میکنند و تاریخچه تکاملی یک تاکسون از آخرین نیای مشترک تا زمان حاضر را نشان میدهند.
-
-
طول شاخهها:
-
در برخی درختها مانند کلادوگرامها، طول شاخهها اختیاری است و معنی خاصی ندارد.
-
در درختهای دیگر مانند فیلوگرامها و درختهای اولترامترک، طول شاخهها اطلاعات منتقل میکند:
-
در فیلوگرام، طول شاخه متناسب با میزان تغییر ژنتیکی است، که بر اساس جایگزینی نوکلئوتیدها یا اسیدهای آمینه اندازهگیری میشود.
-
در درخت اولترامترک، طول شاخه نمایانگر زمان است و همه نوکها در زمان حال قرار دارند.
-
-
نکته مهم: شاخهها محل وقوع فرآیندهای تکاملی مانند جهش، رانش ژنتیکی، انتخاب طبیعی و بازترکیبی هستند. اثرات تجمعی این فرآیندها در واگرایی شاخهها منعکس میشود و شاخهها دینامیک تکاملی را تجسم میکنند.
۳. ریشه (Root)
ریشه یک درخت تبارزایشی نمایانگر آخرین نیای مشترک (MRCA) تمام تاکسونهای درخت است و یک نقطه زمانی و جهتدار برای ترتیب انشعابها فراهم میکند.
-
برای ریشهگذاری درخت، اغلب از اوتگروه (Outgroup) استفاده میشود؛ تاکسونی که خارج از گروه مورد مطالعه است اما به اندازه کافی مرتبط است تا زمینه مقایسه فراهم کند.
-
مثال: در تحلیل روابط نخستیسانان، میتوان یک پستاندار غیرنخستی مانند لمور یا جوندگان را به عنوان اوتگروه وارد کرد.
-
-
بدون ریشهگذاری، درخت بدون ریشه است و تنها روابط را نشان میدهد، نه جهت آنها.
نکته کلیدی: انتخاب ریشه میتواند تفسیر تکاملی را به شدت تحت تأثیر قرار دهد. برای مثال، در بحث درباره اولین شاخههای منشعبشده جانوران، قرار دادن ریشه در موقعیتهای مختلف میتواند فرضیههای متفاوتی درباره منشأ ویژگیهای پیچیده مانند اعصاب و ماهیچهها ایجاد کند.
۴. کلادها (Clades)
کلاد گروهی از موجودات است که یک نیای مشترک و تمام نوادگان آن را شامل میشود. کلادها واحدهای طبیعی زیستشناسی تکاملی هستند و روابط واقعی نسبی را نشان میدهند.
-
کلادها میتوانند در یکدیگر تو در تو باشند و ماهیت سلسلهمراتبی تکامل را نشان دهند.
-
مثال: پستانداران یک کلاد درون مهرهداران، نخستیسانان یک کلاد درون پستانداران، و انسانها یک کلاد درون نخستیسانان هستند.
-
-
کلادها با ویژگیهای مشترک مشتقشده (Synapomorphies) تعریف و نامگذاری میشوند:
-
وجود غدد پستانی → کلاد Mammalia
-
وجود پرها → کلاد Aves
-
نکته مهم: شناخت کلادها امکان بازسازی سیستمهای ردهبندی را به شکلی فراهم میکند که تاریخچه تکاملی را منعکس کند و از گروهبندیهای مصنوعی بر اساس شباهت سطحی فاصله بگیرد.
۵. اوتگروه (Outgroup)
اوتگروه نقش حیاتی در تحلیلهای تبارزایشی دارد و با ارائه نقطه مرجع خارج از گروه مورد مطالعه، امکان ریشهگذاری درخت و تعیین قطبیت تغییرات ویژگیها را فراهم میکند.
-
مثال: در تحلیل روابط مهرهداران، وارد کردن یک لنسلت (Chordate پایهای) به عنوان اوتگروه کمک میکند تشخیص داده شود کدام ویژگیها ابتدایی و کدام مشتق هستند.
-
انتخاب مناسب اوتگروه اهمیت دارد:
-
اگر اوتگروه خیلی دور باشد → ممکن است خطای جذب شاخه طولانی (Long-branch attraction) ایجاد شود.
-
اگر اوتگروه خیلی نزدیک باشد → ممکن است وضوح کافی ارائه نکند.
-
-
اغلب شامل کردن چند اوتگروه برای افزایش اعتبار و robustness تحلیل توصیه میشود.
۶. توپولوژی (Topology)
توپولوژی درخت تبارزایشی به چیدمان گرهها و شاخهها گفته میشود و الگوی ارتباط بین تاکسونها را نشان میدهد. توپولوژی ماهیت فرضیه تکاملی را نشان میدهد و مشخص میکند کدام شاخهها نیای مشترک دارند و ترتیب انشعابها چگونه است.
-
توپولوژی مستقل از طول شاخهها است؛ دو درخت میتوانند توپولوژی یکسان اما طول شاخه متفاوت داشته باشند.
-
تعداد توپولوژیهای ممکن با افزایش تعداد تاکسونها بهشدت افزایش مییابد:
-
۱۰ تاکسون → بیش از ۲ میلیون توپولوژی بدون ریشه
-
۲۰ تاکسون → بیش از 8×10218 \times 10^{21}8×1021 توپولوژی
-
نکته: این افزایش ترکیبیات نشان میدهد که تحلیل تبارزایشی نیاز به روشهای محاسباتی برای جستجوی فضای درختها دارد و یافتن بهترین توپولوژی پشتیبانیشده یکی از چالشهای اصلی است.
۷. ویژگیها و حاشیهنویسیهای اضافی (Additional Features and Annotations)
درختهای تبارزایشی مدرن اغلب شامل حاشیهنویسیهای اضافی هستند که تفسیر آنها را غنی میکنند:
-
مقادیر بوتاسترپ (Bootstrap) یا احتمال پسین (Posterior Probability) در گرهها → نشاندهنده اعتماد آماری به روابط شاخهای
-
نقشهبرداری ویژگیها (Character Mapping): ویژگیهایی مانند ویژگیهای مورفولوژیک، جایگاه اکولوژیکی، یا پراکندگی جغرافیایی روی درخت نمایش داده میشوند.
-
در درختهای مولکولی → شامل میزان جایگزینی، تمایل استفاده از کدون، یا الگوهای ساختاری
-
در درختهای کالیبرهشده بر اساس زمان → گرهها میتوانند دارای تاریخهای برآوردی انشعاب همراه با فاصله اطمینان باشند
نکته مهم: این ویژگیها نشان میدهند که درختهای تبارزایشی صرفاً نمودارهای ثابت نیستند، بلکه فرضیههای پویا هستند
IV. انواع درختهای فیلوژنتیک
درختهای فیلوژنتیک نمایشهای چندمنظورهای از روابط تکاملی هستند و شکل آنها میتواند بسته به ماهیت دادهها، پرسش تحقیق، و روشهای مورد استفاده در ساخت آنها متفاوت باشد. درک انواع درختهای فیلوژنتیک برای تفسیر صحیح بسیار مهم است، زیرا هر شکل بر جنبههای مختلف تاریخ تکاملی تأکید میکند. بهطور کلی، درختهای فیلوژنتیک میتوانند بر اساس ریشهگذاری، نمایش طول شاخهها، نوع اطلاعات تکاملی منتقلشده، و اجماع بین تحلیلهای متعدد دستهبندی شوند. هر نوع دارای مزایای خاص و کاربردهای تحلیلی متفاوت در زیستشناسی تکاملی، سیستماتیک و رشتههای مرتبط است.
1. درختهای ریشهدار (Rooted) در مقابل بیریشه (Unrooted)
تفاوت بین درختهای ریشهدار و بیریشه یکی از مفاهیم اساسی در تحلیل فیلوژنتیک است.
-
درخت بیریشه ریشهای مشخص نمیکند و بنابراین اطلاعاتی درباره جهت زمان تکاملی ارائه نمیدهد. این درخت تنها روابط تاکسونها را از نظر ارتباط و نزدیکی نسبی نشان میدهد و هیچ خط تبار خاصی را بهعنوان اجدادی مشخص نمیکند. درختهای بیریشه اغلب نقطه شروع تحلیلهای فیلوژنتیک هستند، زیرا میتوان آنها را بدون مشخص کردن اوتگروپ (Outgroup) ساخت. سپس با استفاده از اطلاعات خارجی یا تاکسونهای اوتگروپ، ریشهگذاری میشود.
انتخاب بین درخت ریشهدار و بیریشه بر تفسیر تأثیر میگذارد. برای مثال، یک درخت بیریشه میتواند نشان دهد که گونه A و B به هم نزدیکتر هستند تا هر کدام با گونه C، اما مشخص نمیکند که کدام خط تبار ابتدا واگرا شده است. استفاده از اوتگروپ مناسب برای ریشهگذاری این ابهام را برطرف کرده و رویدادهای تکاملی را در یک چارچوب زمانی قرار میدهد.
2. کلادوگرامها، فیلوگرامها و دندروگرامها
درختهای فیلوژنتیک همچنین میتوانند بر اساس نحوه نمایش طول شاخهها و اطلاعات منتقلشده دستهبندی شوند:
-
کلادوگرام (Cladogram):
بر ترتیب انشعابها یا توپولوژی (Topology) تمرکز دارد و میزان تغییر تکاملی را نشان نمیدهد. در کلادوگرام، تمام شاخهها معمولاً به یک طول رسم میشوند و روابط را بر زمان یا فاصله ژنتیکی ترجیح میدهند. کلادوگرامها در کلادیکها (Cladistics) بهویژه مفید هستند، جایی که هدف اصلی شناسایی گروههای مونوفیلتیک (Clades) بر اساس ویژگیهای مشتقشده مشترک است. این نمودارها به پژوهشگران امکان میدهند الگوی تکامل ویژگیها را بدون تداخل با نرخهای واگرایی بررسی کنند. -
فیلوگرام (Phylogram):
طول شاخهها را متناسب با تغییرات تکاملی نشان میدهد، معمولاً بر اساس تفاوتهای ژنتیکی مانند جانشینی نوکلئوتیدها یا جایگزینی اسیدهای آمینه. فیلوگرامها دید دوگانه ارائه میکنند: توپولوژی روابط تاکسونها را نشان میدهد و طول شاخهها نرخهای نسبی تکامل را منتقل میکند. برای مثال، یک شاخه طولانیتر ممکن است نشاندهنده خط تبار با انباشت بیشتر موتاسیونها یا تکامل مولکولی سریعتر باشد. فیلوگرامها در مطالعات مقایسهای، جایی که درک نرخهای متفاوت تغییرات اهمیت دارد، ارزشمند هستند. -
دندروگرام (Dendrogram):
نمودارهای عمومیتر هستند که در زیستشناسی و رشتههای دیگر برای نمایش خوشهبندی سلسلهمراتبی دادهها استفاده میشوند. دندروگرامها ساختار شاخهای درختها را دارند، اما الزاماً درخت تکاملی نیستند و ممکن است نشاندهنده اجداد نباشند. در زیستشناسی مولکولی و اکولوژی، دندروگرامها اغلب برای نشان دادن شباهت بین توالیها، گونهها یا جوامع اکولوژیکی بر اساس معیارهای فاصله استفاده میشوند، حتی اگر فرضیه تکاملی صریحی مدنظر نباشد.
3. درختهای اولترامتر (Ultrametric Trees)
درختهای اولترامتر نوع ویژهای از درختهای فیلوژنتیک هستند که در آن تمام نوکها از ریشه فاصله یکسان دارند. این طراحی نشان میدهد که میزان تغییر تکاملی متناسب با زمان است و برای استفاده از فرضیات ساعت مولکولی (Molecular Clock) بسیار مفید است. با کالیبراسیون گرهها با شواهد فسیلی یا نرخهای موتاسیون شناختهشده، درختهای اولترامتر به پژوهشگران امکان میدهند زمان رویدادهای واگرایی را برآورد و توالی تاریخی تکامل را بازسازی کنند.
این نوع درختها به ویژه در مطالعاتی که الگوهای زمانی گونهزایی، ظهور ویژگیهای جدید، یا ارتباط رویدادهای تکاملی با تغییرات زمینشناسی و اقلیمی را بررسی میکنند، کاربرد دارند. برای مثال، در مطالعه تنوع پستانداران پس از انقراض کرتاسه–پالئوژن (Cretaceous–Paleogene)، درختهای اولترامتر امکان برآورد زمان شکلگیری خطوط تبار اصلی و بررسی همزمانی انفجار گونهزایی با فرصتهای اکولوژیکی ایجادشده توسط انقراض را فراهم میکنند.
4. درختهای اجماعی (Consensus Trees)
در بسیاری از تحلیلهای فیلوژنتیک، چندین درخت ممکن است از یک مجموعه داده ساخته شوند، به ویژه وقتی دادهها مبهم هستند یا روشهای مختلف توپولوژیهای متفاوتی تولید میکنند. درختهای اجماعی برای جمعبندی ویژگیهای مشترک این درختها ساخته میشوند.
انواع درختهای اجماعی شامل:
-
درخت اجماع سخت (Strict Consensus): شامل فقط آن کلادهایی است که در همه درختهای بررسیشده وجود دارند و محافظهکارانهترین برآورد روابط را ارائه میدهد.
-
درخت اجماع اکثریت (Majority-Rule Consensus): شامل کلادهایی است که در اکثریت درختها ظاهر میشوند و تعادلی بین اتفاق نظر سخت و تصویر جامعتر روابط تکاملی ایجاد میکند.
-
درخت اجماع اکثریت گسترده (Extended Majority-Rule Consensus): نسخه توسعهیافتهای است که اطلاعات اضافی در مورد نااطمینانی و الگوهای غیرقطعی ارائه میدهد.
این درختها برای ترکیب نتایج از دادهها، روشها یا تکرارهای بوتاسترپ (Bootstrap) بسیار ارزشمند هستند، زیرا الگوهای تکاملی پایدار و نقاط ابهام را بهطور همزمان نمایش میدهند.
5. درختهای گونهای (Species Trees) در مقابل درختهای ژنی (Gene Trees)
درختهای فیلوژنتیک میتوانند در سطوح زیستشناسی متفاوت ساخته شوند که منجر به تمایز بین درختهای گونهای و درختهای ژنی میشود:
-
درخت گونهای (Species Tree): روابط تکاملی میان گونهها را نشان میدهد و تاریخچه رویدادهای گونهزایی را بازتاب میکند.
-
درخت ژنی (Gene Tree): تاریخچه تکاملی یک ژن یا ناحیه ژنومی خاص را نمایش میدهد. درختهای ژنی ممکن است از درختهای گونهای متفاوت باشند به دلیل فرایندهایی مانند تکثیر ژن، انتقال افقی ژن یا تفکیک نامکمل خطوط تبار (Incomplete Lineage Sorting).
درک تفاوت بین درختهای ژنی و گونهای برای فیلوژنومیکس و ژنومیک مقایسهای حیاتی است، جایی که هدف غالباً بازسازی درخت گونهای با در نظر گرفتن اختلاف بین تاریخچه ژنهای منفرد است.
برای مثال، در گیاهان هیبریدی شدن رایج است و درختهای ژنی ممکن است تاریخچههای پیچیده و شبکهای را نشان دهند، نه فقط خطوط دوتایی. در باکتریها، انتقال افقی ژن میتواند درختهای ژنی ایجاد کند که با تاریخچه ارگانیسم همخوانی ندارند. بنابراین پژوهشگران باید درختهای فیلوژنتیک را در سطح زیستشناسی مناسب تفسیر کرده و تاریخچه خطوط تبار را از تاریخچه ژنی تفکیک کنند.
6. درختهای زمانسنجی شده (Time-Calibrated Trees)
درختهای زمانسنجی شده اطلاعات زمانی صریح را در فیلوژنی ادغام میکنند. این درختها با استفاده از مدلهای ساعت مولکولی، نقاط کالیبراسیون فسیلی، یا روشهای تاریخگذاری دیگر ساخته میشوند. درختهای زمانسنجی شده در زیستشناسی تکاملی و دیرینهشناسی زیستی اهمیت ویژهای دارند، زیرا به پژوهشگران امکان میدهند:
-
زمان شکلگیری کلادهای خاص را برآورد کنند،
-
نرخهای تنوعزیستی را بررسی کنند، و
-
رویدادهای تکاملی را با تغییرات محیطی و زمینشناسی مرتبط کنند.
برای مثال، از درختهای زمانسنجی شده برای مطالعه زمانبندی تنوع پستانداران نسبت به شکستن قارهها، ظهور گیاهان گلدار، یا رویدادهای انقراض جمعی استفاده شده است. این درختها نه تنها ساختار شاخهای، بلکه چارچوب زمانی را نیز ارائه میدهند و قدرت توضیحی داستان تکامل را افزایش میدهند.
7. اشکال ویژه: درختهای شبکهای و رتیکولار (Reticulate and Network Trees)
اکثر درختهای فیلوژنتیک بهطور دقیق دوشاخهای هستند (Bifurcating) و یک سری تقسیمات منظم را نشان میدهند، اما تکامل همیشه به صورت درختی ساده نیست.
-
تکامل رتیکولار (Reticulate Evolution): شامل هیبریدی شدن، انتقال افقی ژن و بازترکیب است که شبکهها را به جای درختهای دوتایی ساده ایجاد میکند.
-
شبکههای فیلوژنتیک (Phylogenetic Networks): نمودارهای تخصصی هستند که این تاریخچههای پیچیده را نمایش میدهند و نقاط رتیکولار (Reticulation Points) را نشان میدهند، جایی که خطوط تبار ادغام یا تبادل ماده ژنتیکی میکنند.
شبکهها به ویژه در تکامل میکروبی، جایی که انتقال افقی ژن شایع است، و در تکامل گیاهان، جایی که هیبریدی شدن میتواند گونههای جدید ایجاد کند، اهمیت دارند. این درختها چارچوب مفهومی فیلوژنتیک را گسترش میدهند و نشان میدهند که تاریخچه واقعی تکامل اغلب پیچیدهتر از مدلهای ساده دوشاخهای است.
بخش پنجم: منابع داده برای استنتاج فیلوژنتیک
تحلیل فیلوژنتیک بهطور اساسی بر دادههایی که سیگنالهای تکاملی را ثبت میکنند متکی است. این دادهها پایهای برای بازسازی روابط میان تاکساها، برآورد زمانهای واگرایی و آزمون فرضیهها درباره تاریخچه تکاملی فراهم میکنند. کیفیت، نوع و گستره دادهها تعیینکننده قابلیت اعتماد و دقت یک درخت فیلوژنتیک است و بنابراین انتخاب و آمادهسازی دادهها یکی از حیاتیترین مراحل در پژوهش فیلوژنتیک محسوب میشود.
طی دههها، فیلوژنتیک از تکیه اصلی بر ویژگیهای مورفولوژیکی به مجموعهای گسترده از دادههای مولکولی و ژنومی شامل DNA، RNA، پروتئینها و توالیهای کل ژنوم تکامل یافته است. هر نوع داده ویژگیها، مزایا و محدودیتهای خاص خود را دارد که بر چگونگی استنتاج روابط تکاملی تأثیر میگذارد.
۱. دادههای مورفولوژیکی
دادههای مورفولوژیکی یا آناتومیکی قبل از ظهور دوران مولکولی، منبع اصلی اطلاعات در فیلوژنتیک بودند. این دادهها شامل ویژگیهای قابل مشاهده مانند ساختارهای اسکلتی، سیستمهای اندامی، شکل بدن و ویژگیهای رشد و نمو هستند. ویژگیهای مورفولوژیکی اغلب بهصورت حالات گسسته کدگذاری میشوند (مثلاً حضور یا عدم حضور یک استخوان خاص، تعداد مهرهها، شکل برگ) و برای استنتاج ریشههای اشتراک تکاملی تحلیل میشوند.
یکی از مزایای دادههای مورفولوژیکی این است که میتوان آنها را برای اندامهای زنده و منقرض شده به کار برد و امکان ادغام تاکساهای فسیلی را در تحلیلهای فیلوژنتیک فراهم میکند. فسیلها اطلاعات زمانی و ساختاری حیاتی ارائه میدهند که از موجودات زنده به تنهایی قابل دسترسی نیست، و این امکان را فراهم میکنند تا ترتیب تکاملی و زمانبندی نوآوریهای کلیدی بازسازی شود. بهعنوان مثال، گنجاندن دوزیستان فسیلی در درختهای فیلوژنتیک به روشن شدن منشأ دوزیستان مدرن و گذار مهرهداران از آب به خشکی کمک کرد.
با این حال، دادههای مورفولوژیکی محدودیتهایی نیز دارند. تکامل همگرا میتواند شباهتهایی را در تاکساهای نامرتبط ایجاد کند که در صورت عدم ارزیابی دقیق، به تفسیر نادرست روابط منجر میشود. علاوه بر این، ویژگیهای مورفولوژیکی میتوانند در کدگذاری ذهنی یا متکی بر قضاوت شخصی باشند و در تعریف حالات ویژگی یا ارزیابی همولوژی ابهام ایجاد کنند. با وجود این چالشها، دادههای مورفولوژیکی همچنان غیرقابل جایگزین هستند، بهویژه در مطالعات مربوط به گونههای منقرض شده یا تاکساهایی که دادههای مولکولی برای آنها موجود نیست.
۲. دادههای مولکولی: DNA، RNA و پروتئینها
توالیهای مولکولی منبعی فراوان و بسیار اطلاعاتی برای استنتاج فیلوژنتیک فراهم میکنند. توالیهای DNA، شامل ژنومهای هستهای، میتوکندریایی و کلروپلاستی، امکان مقایسه مستقیم ماده ژنتیکی بین تاکساها را فراهم میکنند. توالیهای RNA، بهویژه RNA ریبوزومی، به دلیل حفظ تکاملی و حضور در تمام حوزههای زندگی نقش کلیدی دارند. توالیهای پروتئین، که از DNA استنتاج میشوند یا مستقیماً توالییابی میشوند، اطلاعات اضافی ارائه میدهند و برای بررسی روابط تکاملی عمیق بسیار مفید هستند.
مزایای دادههای مولکولی نسبت به دادههای مورفولوژیکی عبارتند از:
-
ارائه اطلاعات کمی که میتوان بهطور دقیق با مدلهای آماری تکامل توالیها تحلیل کرد.
-
فراوانی و قابلیت ذخیره دیجیتال، که امکان تحلیلهای سراسری و بزرگمقیاس را فراهم میکند.
-
امکان بررسی توالیهای آهستهتکاملی برای تحلیل واگراییهای عمیق و توالیهای سریعتکاملی برای تحلیل تقسیمات اخیر.
با این وجود، دادههای مولکولی نیز با چالشهایی مواجه هستند. همولوژی ناقص (homoplasy) ناشی از چندین جایگزینی در یک سایت میتواند روابط واقعی را بهویژه در نواحی سریعتکاملی مخدوش کند. همچنین، انتقال افقی ژن، تکثیر ژن و نارسایی در جدایی تبارها میتوانند باعث ناسازگاری بین درختهای ژن و درخت گونهها شوند. برای کاهش این مشکلات، فیلوژنتیکدانان اغلب از چندین ژن یا دادههای سطح ژنوم استفاده میکنند و روشهایی را به کار میبرند که ناهمگنی درختهای ژن را مدنظر قرار میدهند.
۳. دادههای ژنومی و فیلوژنومی
ظهور توالییابی پرسرعت دادههای فیلوژنتیک را از چند ژن به کل ژنومها گسترش داده است و زمینه را برای فیلوژنومیک (Phylogenomics) فراهم کرده است. روشهای فیلوژنومیک دادههای صدها تا هزاران لوکوس را در چندین گونه یکپارچه میکنند و قدرت آماری و دقت تحلیلهای فیلوژنتیک را بهطور چشمگیری افزایش میدهند. این رویکرد بهویژه برای حل مسائل دشوار مانند تشعشعات سریع، واگراییهای عمیق یا روابط میان شاخههایی با تفاوت مورفولوژیکی محدود ارزشمند است.
دادههای کل ژنوم اطلاعاتی درباره ناحیههای کدکننده و غیرکدکننده، واریانتهای ساختاری و سینتنی (ترتیب ژنها) فراهم میکنند و دیدی چندلایه از تاریخچه تکاملی ارائه میدهند. دادههای ژنوممحور همچنین امکان استفاده از روشهای پیشرفته برای مدلسازی فرآیندهای تکاملی، از جمله نرخهای جایگزینی، فشارهای انتخاب و الگوهای بازترکیب را فراهم میکنند. فیلوژنومیک در روشن کردن درخت حیات، حل روابط جنجالی بین شاخههای اصلی جانوران و ارائه دیدگاههایی درباره منشأ گیاهان، قارچها و شاخههای میکروبی نقش کلیدی داشته است.
۴. دادههای ترنسکریپتومیک و پروتئومیک
دادههای ترنسکریپتومیک که از توالییابی RNA بهدست میآیند، بخش بیانشده ژنوم را نشان میدهند و میتوانند سیگنال فیلوژنتیک مکمل DNA ارائه دهند. زیرا RNA توالیهای کدکننده و الگوهای بیان ژنها را منعکس میکند، این دادهها بهویژه برای تاکساهایی که توالییابی کل ژنوم چالشبرانگیز است مفید هستند. تحلیلهای ترنسکریپتومیک امکان شناسایی ژنهای اورتوگال برای بازسازی فیلوژنتیک و اطلاع از تکامل عملکردی را فراهم میکنند و نشان میدهند کدام ژنها یا مسیرها بهطور متفاوت تکامل یافتهاند.
دادههای پروتئومیک، که توالیهای آمینواسیدی پروتئینهای بیانشده را نشان میدهند، لایه مکمل دیگری ارائه میکنند. پروتئینها تحت محدودیتهای عملکردی قرار دارند و مقایسه توالیهای پروتئینی حفظشده برای استنتاج روابط تکاملی عمیق ارزشمند است. هر دو داده ترنسکریپتومیک و پروتئومیک نیازمند حاشیهنویسی و همترازی دقیق هستند اما برای استنتاج فیلوژنتیک، بهویژه در ارگانیسمهای غیرمدل اطلاعات غنی فراهم میکنند.
۵. دادههای میکروبی و متاژنومیک
فیلوژنتیک میکروبی چالشها و فرصتهای منحصربهفرد دارد، زیرا انتقال افقی ژن رایج است و بسیاری از میکروارگانیسمها قابل کشت نیستند. روشهای متاژنومیک مدرن اجازه میدهند تا DNA محیطی مستقیماً توالییابی شده، ژنومهای میکروبی بازسازی شوند و تاکساهای غیرقابل کشت در درختهای فیلوژنتیک قرار گیرند. ژنهای شاخص مانند 16S rRNA در باکتریها بهطور گسترده برای شناسایی تاکساها و استنتاج روابط استفاده میشوند. تحلیل فیلوژنتیک جوامع میکروبی اطلاعاتی درباره تعاملات اکولوژیکی، جغرافیای زیستی و دینامیک تکاملی ارائه میدهد.
دادههای متاژنومیک نیازمند خطوط کاری بیوانفورماتیکی تخصصی برای مونتاژ توالیها، شناسایی ژنهای اورتوگال و مدیریت ژنومهای ناقص یا خرد شده هستند. با وجود این چالشها، متاژنومیک درک ما از تنوع میکروبی را متحول کرده و دامنه تحلیلهای فیلوژنتیک را به اکثر حیات که بهراحتی کشت نمیشود گسترش داده است.
۶. ادغام دادههای مورفولوژیکی و مولکولی
رویکردی که به طور فزاینده استفاده میشود، ادغام دادههای مورفولوژیکی و مولکولی است، بهویژه در مطالعاتی که شامل تاکساهای زنده و منقرض شده هستند. با ترکیب شواهد فسیلی با دادههای ژنومی، پژوهشگران میتوانند درختهایی تولید کنند که هم عمق تاریخی و هم جزئیات ژنتیکی را در بر داشته باشند. این ادغام امکان زمانبندی دقیقتر رویدادهای واگرایی، وضوح بهتر گرههای عمیق و درک دقیقتر تکامل ویژگیها را فراهم میکند.
تحلیلهای ترکیبی نیازمند مدیریت دقیق دادههای گمشده، کدگذاری ویژگیها و انتخاب مدل هستند. روشهایی مانند تحلیل شواهد جامع (total evidence) یا رویکردهای تاریخگذاری نوک (tip-dating)، دادههای مورفولوژیکی و مولکولی را در یک چارچوب واحد ترکیب میکنند و دیدگاهی جامع از تاریخچه تکاملی ارائه میدهند. این رویکردها فلسفه مدرن فیلوژنتیک را نشان میدهند، جایی که چندین منبع شواهد برای ساخت فرضیههای محکم و قابل آزمون استفاده میشوند.
۷. منابع داده دیگر
فراتر از دادههای کلاسیک مورفولوژیکی و مولکولی، استنتاج فیلوژنتیک میتواند انواع مختلفی از اطلاعات را در بر گیرد. ویژگیهای رفتاری، مشخصات اکولوژیکی و ویژگیهای رشد و نمو میتوانند سیگنال فیلوژنتیک ارائه دهند، بهویژه زمانی که با سایر دادهها تحلیل شوند. در فیلوژنتیک فرهنگی، زبانها، فناوریها یا شیوههای اجتماعی مشابه ویژگیهای بیولوژیکی در نظر گرفته میشوند و درختهایی ساخته میشوند که تکامل فرهنگ انسانی را منعکس کنند.
انتخاب و کدگذاری دقیق این دادهها ضروری است، زیرا ویژگیهای غیرژنتیکی ممکن است تحت تأثیر همگرایی، انتقال افقی یا تکامل سریع قرار گیرند که ساخت درخت را مختل میکند. با این حال، زمانی که بهدرستی تحلیل شوند، این دادهها ابعاد اضافی برای درک الگوها و فرآیندهای تکاملی فراهم میکنند.
بخش ششم: روشهای ساخت درخت فیلوژنتیک
ساخت یک درخت فیلوژنتیک شامل تحلیل نظاممند دادههای زیستی برای استنتاج روابط تکاملی میان تاکساها است. طی سالها، روشهای متعددی توسعه یافتهاند که بازتابدهنده پیشرفت در قدرت محاسباتی، نظریه آماری و دسترسی به دادههای مولکولی هستند. ساخت درخت فیلوژنتیک در واقع یک تمرین در تولید فرضیه است: هر درخت یک مدل قابل آزمون از تاریخچه تکاملی بر اساس دادهها و روششناسی انتخابشده است. روشهای ساخت درخت را میتوان به سه دسته کلی تقسیم کرد: روشهای مبتنی بر فاصله، روشهای مبتنی بر ویژگی (character-based) و روشهای آماری یا مبتنی بر مدل، که هر کدام اصول، مزایا و محدودیتهای خاص خود را دارند. درک این روشها برای انتخاب چارچوب تحلیلی مناسب و تفسیر صحیح درختهای حاصل ضروری است.
۱. روشهای مبتنی بر فاصله (Distance-Based Methods)
روشهای مبتنی بر فاصله یکی از اولین و شهودیترین رویکردها برای بازسازی درخت فیلوژنتیک هستند. این روشها با کمیسازی اختلاف میان جفتهای تاکساها آغاز میشوند، معمولاً به شکل یک ماتریس فاصله، و سپس با استفاده از الگوریتمها، درختی ساخته میشود که بهترین بازنمایی از این فاصلهها باشد. معیارهای فاصله رایج شامل فاصله ژنتیکی، جایگزینی نوکلئوتیدی یا اختلافات مورفولوژیکی هستند.
الگوریتم UPGMA (Unweighted Pair Group Method with Arithmetic Mean) یک روش کلاسیک مبتنی بر فاصله است. UPGMA درخت ریشهدار میسازد و تاکساها را به صورت تکراری براساس کوچکترین فاصلهها خوشهبندی میکند و فرض میکند که ساعت مولکولی برقرار است؛ یعنی نرخ تکامل در همه شاخهها یکسان است. اگرچه UPGMA از نظر محاسباتی کارآمد و مفهومی ساده است، اما فرض نرخ تکامل برابر در همه شاخهها میتواند مشکلساز باشد و منجر به ناهمخوانی در توپولوژی درخت شود، بهویژه وقتی نرخها در میان شاخهها متفاوت باشند.
روش دیگر مبتنی بر فاصله، الگوریتم Neighbor-Joining (NJ) است که درخت بدون ریشه تولید میکند و فرض ساعت مولکولی ندارد. NJ جفتهای تاکسا یا خوشههایی را شناسایی میکند که حداقل طول شاخه کل را در هر مرحله داشته باشند و درخت را به تدریج میسازد. به دلیل امکان تطبیق با نرخهای متغیر تکامل، NJ از UPGMA انعطافپذیرتر است و در فیلوژنتیک مولکولی برای بازسازی روابط از توالیهای DNA یا پروتئین بهطور گسترده استفاده میشود. روشهای مبتنی بر فاصله سریع و مناسب برای دادههای بزرگ هستند، اما اطلاعات پیچیده توالی را به یک شاخص خلاصه کاهش میدهند که ممکن است منجر به از دست رفتن سیگنال فیلوژنتیک شود.
۲. روشهای مبتنی بر ویژگی (Character-Based Methods)
روشهای مبتنی بر ویژگی به تحلیل مستقیم هر ویژگی—نوکلئوتیدها، اسیدهای آمینه یا ویژگیهای مورفولوژیکی—میپردازند و آنها را به فاصلهها خلاصه نمیکنند. هدف این روشها یافتن توپولوژی درختی است که بهترین توضیح برای توزیع مشاهدهشده ویژگیها در تاکساها ارائه دهد و اغلب از اصل صرفهجویی یا پارسیمونی (parsimony) استفاده میکنند.
روش Maximum Parsimony (MP) درختی را شناسایی میکند که کمترین تغییرات تکاملی را برای توضیح حالتهای مشاهدهشده ویژگیها نیاز دارد. به عبارت دیگر، MP همولوژی ناقص (homoplasy) را به حداقل میرساند و فرض میکند که تکامل مسیر سادهتر را دنبال میکند. پارسیمونی در فیلوژنتیک مورفولوژیکی و مولکولی بهویژه زمانی که نرخهای تکاملی نسبتا یکنواخت و همگرایی محدود است، کاربرد دارد. MP مفهومی جذاب و تولید درختهای قابل تفسیر آسان دارد، اما میتواند نسبت به جذب شاخه بلند (long-branch attraction) حساس باشد، یعنی زمانی که شاخههای با تکامل سریع به اشتباه کنار هم گروهبندی شوند.
روشهای مبتنی بر ویژگی همچنین شامل رویکردهای Minimum Evolution هستند که اصول تحلیل ویژگیها را با بهینهسازی طول شاخهها ترکیب میکنند. این روشها با ادغام اطلاعات ویژگیها و بهینهسازی طول شاخه، توازنی بین سادگی و وفاداری به دادهها برقرار میکنند.
۳. روشهای آماری مبتنی بر مدل (Model-Based Statistical Methods)
پیشرفت در نظریه تکامل مولکولی منجر به توسعه روشهای مبتنی بر مدل شده است، که مدلهای صریح تغییر تکاملی را در نظر میگیرند. این روشها شامل Maximum Likelihood (ML) و Bayesian Inference (BI) هستند، که هر دو امکان ارزیابی آماری دقیق فرضیههای درخت را فراهم میکنند.
روش Maximum Likelihood توپولوژی درخت و طول شاخهها را تخمین میزند تا احتمال مشاهده دادههای موجود تحت یک مدل خاص تکامل توالی را بیشینه کند. مدلها ممکن است شامل نرخ جایگزینی، نسبت انتقال/ترانسورژن و ناهمگنی نرخ در میان سایتها باشند. روشهای ML محاسباتی سنگین هستند اما وقتی مدلها مناسب باشند، درختهای بسیار دقیقی ارائه میدهند. این روشها همچنین امکان مقایسه آماری فرضیههای جایگزین با استفاده از آزمون نسبت احتمال یا معیارهای اطلاعاتی را فراهم میکنند.
روشهای Bayesian Inference با گنجاندن اطلاعات پیشین درباره پارامترهای درخت و ادغام عدم قطعیتها با چارچوبهای احتمالاتی، ML را توسعه میدهند. روشهای Bayesian یک توزیع احتمال پسین درختها تولید میکنند که سطح اطمینان برای هر کلاد را ارائه میدهد. الگوریتمهای Markov Chain Monte Carlo (MCMC) معمولا برای نمونهگیری از این توزیع استفاده میشوند. روشهای Bayesian انعطافپذیری بالایی دارند و امکان گنجاندن مدلهای پیچیده، ساعت مولکولی انعطافپذیر و نقاط کالیبراسیون فسیلی را فراهم میکنند. این روشها بهویژه در مطالعات فیلوژنومیک و تحلیلهایی که نیازمند سنجش دقیق عدم قطعیت هستند به کار میروند.
۴. بوتاسترپینگ و شاخصهای پشتیبانی (Bootstrapping and Support Measures)
بدون توجه به روش استفادهشده برای ساخت درخت، ارزیابی استحکام روابط استنتاجشده ضروری است. بوتاسترپینگ یک تکنیک آماری متداول است که شامل نمونهگیری مجدد دادهها با جایگزینی برای تولید چندین مجموعه داده شبهتکراری میشود. برای هر نمونه، درخت ساخته میشود و نسبت نمونههایی که از یک کلاد خاص پشتیبانی میکنند محاسبه میشود که مقدار بوتاسترپ نامیده میشود. مقادیر بالای بوتاسترپ نشاندهنده پشتیبانی قوی و مقادیر پایین نشاندهنده عدم اطمینان یا سیگنال متناقض در دادهها است.
شاخصهای پشتیبانی دیگر شامل احتمال پسین در تحلیل Bayesian و شاخصهای decay در تحلیل پارسیمونی هستند. این شاخصها به پژوهشگران اجازه میدهند قابلیت اعتماد روابط خاص در درخت را تفسیر کنند و مناطق نیازمند داده یا تحلیل بیشتر را شناسایی کنند.
۵. مدیریت ناسازگاری درخت ژن–درخت گونه (Gene Tree–Species Tree Discordance)
در فیلوژنتیک مولکولی، درختهای ژن منفرد ممکن است با درخت گونه واقعی متفاوت باشند، به دلیل تکثیر ژن، انتقال افقی ژن، نارسایی در جدایی تبارها یا بازترکیب. مدیریت این ناسازگاری بسیار حیاتی است، بهویژه در مطالعات فیلوژنومیک با تعداد زیادی لوکوس. روشهایی مانند تخمین درخت گونه مبتنی بر کوآلسنت (coalescent-based) بهطور صریح فرآیندهای تصادفی ایجاد ناسازگاری را مدلسازی میکنند و اطلاعات چندین درخت ژن را ادغام میکنند تا درخت گونه اصلی را استنتاج کنند. ابزارهایی مانند ASTRAL، BEST و BEAST این رویکردها را پیادهسازی میکنند و چارچوبی برای استنتاج مستحکم حتی در مواجهه با تضاد در تاریخچه ژنها فراهم میکنند.
۶. نرمافزارها و ابزارهای محاسباتی (Software and Computational Tools)
تحلیلهای فیلوژنتیک مدرن به شدت به ابزارهای محاسباتی برای پیادهسازی این روشها وابسته هستند. نرمافزارهای رایج شامل MEGA، PAUP، RAxML، MrBayes و IQ-TREE* هستند که هر کدام الگوریتمها، مدلها و رابطهای کاربری متفاوت ارائه میدهند. پیشرفت در محاسبات با کارایی بالا امکان تحلیل دادههای سطح ژنوم و استفاده از مدلهای پیچیده با در نظر گرفتن ناهمگنی نرخ، تمایل کدون و فرآیندهای تکاملی پیچیده را فراهم میکند.
این ابزارهای نرمافزاری نه تنها ساخت درخت را تسهیل میکنند، بلکه تصویرسازی، حاشیهنویسی و ارزیابی آماری را نیز پشتیبانی میکنند. آنها به کاربران امکان میدهند توپولوژیهای جایگزین را بررسی کرده، شاخصهای اطمینان را ارزیابی و دادههای مولکولی و مورفولوژیکی را ادغام کنند. دسترسی به نرمافزارهای قدرتمند و کاربرپسند، تحلیل فیلوژنتیک را در دسترس طیف وسیعی از پژوهشگران قرار داده و امکان استفاده از روشهای پیشرفته را فراهم کرده است.
۷. ملاحظات و بهترین شیوهها (Considerations and Best Practices)
ساخت مؤثر درخت فیلوژنتیک نیازمند توجه دقیق به چندین عامل است، از جمله:
-
انتخاب تاکساها: اطمینان از نمونهبرداری نماینده از کلاد مورد نظر.
-
انتخاب ویژگیها یا لوکوسهای مناسب: اجتناب از توالیهای گمراهکننده یا غیر اورتوگال.
-
انتخاب مدل: تطبیق روشهای تحلیلی با زمینه زیستی و تکاملی دادهها.
همچنین، تفسیر درختها باید با نقد و بررسی همراه باشد، با توجه به عدم قطعیت، سوگیریهای احتمالی و محدودیتهای ذاتی هر مجموعه داده.
ترکیب چند روش و منبع داده اغلب منجر به نتایج مستحکمتر میشود. بهعنوان مثال، استفاده از روشهای مبتنی بر فاصله برای تولید درختهای اولیه و سپس اصلاح آنها با روشهای مبتنی بر ویژگی یا مبتنی بر مدل میتواند کارایی محاسباتی و دقت را بهینه کند. ادغام شواهد مولکولی، مورفولوژیکی و فسیلی نیز استنتاج را تقویت میکند و به پژوهشگران امکان میدهد درختهایی بسازند که هم دادهمحور و هم از نظر زیستی معنادار باشند.
بخش هفتم: کاربردهای درختهای فیلوژنتیک
درختهای فیلوژنتیک تنها ساختارهای نظری نیستند؛ بلکه ابزارهای عملی هستند که در طیف وسیعی از رشتههای زیستی اطلاعات عمیق و مفیدی ارائه میدهند. با روشن کردن روابط میان موجودات زنده، ژنها یا جمعیتها، درختهای فیلوژنتیک به پژوهشگران امکان میدهند پرسشهای اساسی در تکامل، اکولوژی، ژنومیک، پزشکی و حفاظت از تنوع زیستی را بررسی کنند. انعطافپذیری تحلیلهای فیلوژنتیک در این است که الگوهای تاریخی نسب را با پدیدههای زیستی معاصر پیوند میدهد و این امر آن را به محور تحقیقات بنیادی و کاربردی تبدیل کرده است.
۱. درک روابط تکاملی (Understanding Evolutionary Relationships)
کاربرد اصلی و آشکار درختهای فیلوژنتیک، روشن کردن روابط تکاملی میان تاکساها است. درختها به زیستشناسان اجازه میدهند مشاهده کنند کدام موجودات دارای اجداد مشترک هستند، زمان نسبی رخدادهای جدایی و ساختار سلسلهمراتبی تاریخچه تکاملی چیست. برای مثال، مکان انسان در درخت نخستیسانان، نزدیکی ما به شامپانزهها و بونوبوها را نشان میدهد، در حالی که شاخههای دورتر، انسان را به دیگر پستانداران و مهرهداران متصل میکنند.
علاوه بر نمایش وابستگیها، درختهای فیلوژنتیک چارچوبی برای تفسیر توالی رخدادهای تکاملی فراهم میکنند. با شناسایی گرههای اجدادی و دنبال کردن جدایی شاخهها، پژوهشگران میتوانند ظهور نوآوریهای کلیدی مانند پرها در پرندگان، چندسلولی بودن در یوکاریوتها یا فتوسنتز در گیاهان را بازسازی کنند. بنابراین، درختها ستون فقرات درک الگوهای ماکروتکاملی هستند و زمینهای ساختاری فراهم میکنند که پیچیدگی تاریخ حیات در آن تحلیل و منتقل شود.
۲. ژنومیک تطبیقی و تکامل مولکولی (Comparative Genomics and Molecular Evolution)
درختهای فیلوژنتیک برای ژنومیک تطبیقی ضروری هستند، جایی که تاریخچه تکاملی ژنها، پروتئینها و ژنومها بررسی میشود. با نقشهبرداری تغییرات ژنتیکی روی درختها، پژوهشگران میتوانند اورتوگها (ژنهایی که به دلیل اسپسیاسیون از هم جدا شدهاند) و پارالوگها (ژنهایی که به دلیل تکثیر از هم جدا شدهاند) را شناسایی کنند، حفظ عملکرد ژنها را از واگرایی تفکیک کنند و نیروهای انتخابی تأثیرگذار بر توالیها را استنتاج کنند.
برای مثال، در مطالعه تکامل خانوادههای ژنی مانند هموگلوبین یا سیتوکروم P450، تحلیل فیلوژنتیک میتواند نشان دهد چگونه تکثیر ژنها باعث تنوع عملکردی شده است. درختها همچنین امکان شناسایی عناصر تکاملی حفظشده را فراهم میکنند، که اغلب ناحیههای عملکردی مهم ژنوم هستند. مطالعات مقایسهای تکامل مولکولی بر فیلوژنیها برای قرار دادن تنوع ژنتیکی در زمینه تاریخی تکیه دارند و میتوانند اجداد مشترک را از تکامل همگرا یا انتقال افقی ژن تفکیک کنند.
تحلیل فیلوژنتیک همچنین بازسازی توالیهای اجدادی را امکانپذیر میکند. با استنتاج ترکیب ژنتیکی ژنها یا پروتئینهای باستانی، پژوهشگران میتوانند خواص بیوشیمیایی مولکولهای اجدادی را بررسی، تکامل عملکرد آنزیمی را مطالعه و فرضیههای مربوط به سازگاری و نوآوری مولکولی را آزمایش کنند.
۳. بیوجغرافیا و اکولوژی تاریخی (Biogeography and Historical Ecology)
درختهای فیلوژنتیک برای مطالعات بیوجغرافیایی که توزیع مکانی و زمانی موجودات را بررسی میکنند، بسیار ارزشمند هستند. با ترکیب فیلوژنیها با دادههای جغرافیایی، پژوهشگران میتوانند حرکات تاریخی گونهها، رویدادهای استقرار و الگوهای تنوعزایی را استنتاج کنند.
برای مثال، تحلیل فیلوژنتیک پرندگان بدون توان پرواز مانند شترمرغ، امو و کیوی، زمانبندی جدایی قارهها و رویدادهای پراکنش که توزیع آنها را شکل داده است، روشن کرده است. به همین ترتیب، درختهای فیلوژنتیک شاخههای گیاهی میتوانند الگوهای مهاجرت، تأثیر تغییرات اقلیمی و ترکیب تاریخی فلور منطقهای را آشکار کنند. چنین مطالعاتی تاریخچه تکاملی را با اکولوژی پیوند میدهند و بینشهایی درباره عوامل مؤثر بر غنای گونهای، اندمیک بودن و ترکیب جامعه ارائه میدهند.
۴. زیستشناسی حفاظت (Conservation Biology)
درختهای فیلوژنتیک نقش حیاتی در زیستشناسی حفاظت دارند و اولویتبندی برای حفظ تنوع زیستی را هدایت میکنند. با کمیسازی تمایز تکاملی (evolutionary distinctiveness)، درختها به حفاظتگران اجازه میدهند گونههایی که نماینده شاخههای تکاملی منحصر به فرد هستند را شناسایی کنند. حفظ چنین گونههایی، گستره تاریخ تکاملی را حفظ میکند، نه صرفاً شمار گونهها.
برای مثال، توآتارا در نیوزیلند نماینده شاخهای است که بیش از ۲۰۰ میلیون سال پیش از سایر خزندگان جدا شده است. حفاظت از آن حیاتی است زیرا از دست دادن آن یک شاخه کامل از درخت زندگی مهرهداران را حذف میکند. به همین ترتیب، شاخصهای تنوع فیلوژنتیک برای طراحی مناطق حفاظتشده استفاده میشوند تا اطمینان حاصل شود که تلاشهای حفاظتی هم تنوع تاکسونومی و هم تاریخچه تکاملی را پوشش میدهد.
درختهای فیلوژنتیک همچنین در مدیریت جمعیتهای در خطر با آشکارسازی تنوع مخفی (cryptic diversity)—شاخههای تکاملی متفاوت که ممکن است از نظر مورفولوژیکی قابل مشاهده نباشند—کمک میکنند. این اطلاعات به اولویتبندی جمعیتها برای حفاظت و هدایت برنامههای پرورشی برای حفظ تنوع ژنتیکی کمک میکند.
۵. اپیدمیولوژی و بهداشت عمومی (Epidemiology and Public Health)
در زمینه اپیدمیولوژی، درختهای فیلوژنتیک برای ردیابی تکامل و گسترش پاتوژنها ضروری هستند. درختهای ساختهشده از توالیهای ویروسی یا باکتریایی میتوانند مسیرهای انتقال، منابع شیوع و ظهور مقاومت دارویی را نشان دهند.
برای مثال، در طول پاندمی COVID-19، تحلیل فیلوژنتیک توالیهای SARS-CoV-2 به پژوهشگران امکان داد گسترش واریانتها، زمان ورود به مناطق مختلف و الگوهای تکامل ویروس را ردیابی کنند. رویکردهای مشابه برای HIV، آنفلوآنزا و ابولا نیز استفاده شده و اطلاعات حیاتی برای مداخلات بهداشت عمومی و طراحی واکسن فراهم کرده است.
درختهای فیلوژنتیک همچنین درک واگیریهای زئونوتیک را تسهیل میکنند و نشان میدهند کدام مخازن حیوانی ویروسهایی دارند که ممکن است انسان را آلوده کنند. این دانش، راهبری نظارت و سیاستهای پیشگیری از پاندمیهای آینده را هدایت میکند.
۶. کشاورزی و مطالعات اهلیسازی (Agriculture and Domestication Studies)
درختهای فیلوژنتیک کاربردهای مهمی در کشاورزی دارند، به ویژه در درک منشاء و تنوع گونههای زراعی و دامها. با بازسازی تاریخچه تکاملی گیاهان و حیوانات اهلیشده، پژوهشگران میتوانند جدهای وحشی را شناسایی، تاریخچه انتخاب و پرورش را دنبال و منابع ژنتیکی برای بهبود محصول را کشف کنند.
برای مثال، تحلیل فیلوژنتیک تاریخچه پیچیده اهلیسازی گندم، ذرت و برنج را نشان داده است، منشاء جغرافیایی شاخههای مختلف و رویدادهای هیبریداسیون که رقمهای مدرن را شکل دادهاند، مشخص کرده است. در دامپروری، درختهای ساختهشده از DNA میتوکندری یا ژنوم کامل به برنامههای پرورشی، حفاظت ژنتیکی و مدیریت مقاومت بیماریها کمک میکنند.
ابزارهای فیلوژنتیک همچنین یافتن اقوام وحشی با ویژگیهای مطلوب مانند مقاومت به خشکی یا بیماری را تسهیل میکنند و امکان ترکیب هدفمند این ویژگیها در خطوط اهلی را فراهم میکنند. با درک روابط تکاملی، دانشمندان کشاورزی میتوانند حداکثر تنوع ژنتیکی را حفظ و در عین حال بهرهوری، مقاومت و پایداری محصولات را بهبود دهند.
۷. علوم قضایی و کاربردهای حقوقی (Forensic Science and Legal Applications)
درختهای فیلوژنتیک به طور فزایندهای در علوم قضایی به کار میروند، جایی که توالیهای مولکولی برای بازسازی روابط میان نمونههای زیستی استفاده میشوند. در پروندههای کیفری و مدنی، درختها میتوانند به ثابت کردن ارتباط میان افراد، شناسایی منابع ماده ژنتیکی یا ردیابی منشأ عوامل عفونی کمک کنند.
برای مثال، تحلیل فیلوژنتیک در موارد انتقال HIV استفاده شده است، جایی که درختهای ساختهشده از توالیهای ویروس میتوانند نشان دهند آیا یک فرد ویروس را به فرد دیگری منتقل کرده است یا خیر. چنین کاربردهایی نیازمند اعتبارسنجی آماری دقیق و تفسیر محتاطانه هستند تا قابلیت اعتماد قانونی داشته باشند، اما اهمیت عملی تحلیل تکاملی در زمینههای اجتماعی و حقوقی را نشان میدهند.
۸. آموزش و درک عمومی از تکامل (Education and Public Understanding of Evolution)
فراتر از پژوهش، درختهای فیلوژنتیک ابزارهای آموزشی قدرتمندی هستند که به دانشآموزان و عموم مردم کمک میکنند روابط تکاملی را مشاهده و اتحاد و تنوع حیات را درک کنند. درختها میتوانند مفاهیمی مانند اجداد مشترک، تابش انطباقی و تکامل همگرا را نمایش دهند و فرآیندهای انتزاعی تکاملی را ملموس و قابل فهم کنند.
در کلاسها، درختهای دیجیتال و تعاملی امکان میدهند دانشآموزان درخت زندگی را پویا کاوش کنند، شاخهها را دنبال کنند، ژنومها را مقایسه کنند و ویژگیها را به تاریخچه تکاملی مرتبط کنند. با ارائه چارچوبی ساختاری برای درک تنوع زیستی، درختهای فیلوژنتیک به سواد علمی و درک عمومی تکامل کمک میکنند.
بخش هشتم: محدودیتها و چالشهای درختهای فیلوژنتیک
درختهای فیلوژنتیک ابزارهای قدرتمندی برای درک روابط تکاملی هستند، اما بدون محدودیت و چالش نیستند. ساخت و تفسیر فیلوژنیها مستلزم فرضیات متعدد، انتخابهای روششناختی و منابع عدم قطعیت است که همه میتوانند دقت و قابلیت اعتماد درختهای حاصل را تحت تأثیر قرار دهند. شناخت این محدودیتها برای تحلیل دقیق، تفسیر درست و انتقال مؤثر یافتههای تکاملی ضروری است. این چالشها شامل انتخاب و کیفیت دادهها، فرضیات مدل، محدودیتهای روششناختی و پیچیدگیهای زیستی ذاتی تکامل میشوند.
۱. دادههای ناقص یا دارای سوگیری (Incomplete or Biased Data)
یکی از اساسیترین چالشها در تحلیل فیلوژنتیک، ناقص بودن یا سوگیری دادههای موجود است. دادههای مورفولوژیکی ممکن است شخصیتهای حیاتی برخی تاکساها را نداشته باشند، بهویژه در موجودات فسیلی که حفظ آنها ناقص است. دادههای مولکولی نیز میتوانند به دلیل توالیهای ناقص، پوشش پایین یا خطاهای توالییابی محدود شوند. علاوه بر این، نمونهبرداری تاکساها ممکن است عمدی یا غیرعمدی سوگیری داشته باشد و در نتیجه درختهایی ایجاد شود که روابط تکاملی واقعی را بهدرستی نشان ندهند.
دادههای ناقص میتوانند منجر به کاهش وضوح درخت، وجود چندین توپولوژی به طور برابر محتمل، یا خطاهای سیستماتیک شوند. برای مثال، در فیلوژنتیک میکروبی، اکثر گونهها هنوز کشت نشده و توالییابی نشدهاند، بنابراین درختهای ساختهشده از توالیهای موجود ممکن است شاخههای تکاملی اصلی را نادیده بگیرند. استراتژیهایی مانند افزایش نمونهبرداری تاکساها، ترکیب انواع دادههای مختلف یا استفاده از روشهای مقاوم به دادههای ناقص میتوانند این چالشها را کاهش دهند، اما مشکل همچنان محدودیت پایدار باقی میماند.
۲. همریختی و تکامل همگرا (Homoplasy and Convergent Evolution)
همریختی (homoplasy)—شباهت در شخصیتها که ناشی از جد مشترک نیست—چالش مهم دیگری است. تکامل همگرا، تکامل موازی و بازگشتها میتوانند شباهتهای مورفولوژیکی یا مولکولی میان تاکساهای غیرمرتبط ایجاد کنند و استنتاج فیلوژنتیک را گمراه کنند. برای مثال، تکامل بالها در خفاشها و پرندگان نمونه کلاسیک همگرایی است: درختی که فقط بر اساس وجود بالها ساخته شود، این شاخههای دور از هم را به اشتباه در یک گروه قرار میدهد.
در دادههای مولکولی، همریختی ممکن است به دلیل چندین جایگزینی در یک موقعیت، اشباع نواحی به سرعت در حال تکامل یا تعصب کدون ایجاد شود. شناسایی و کنترل همریختی ضروری است و اغلب نیازمند مدلهای پیچیده تکامل توالی، انتخاب دقیق شخصیتها یا فیلتر کردن موقعیتهای بسیار متغیر است. نادیده گرفتن همریختی میتواند منجر به توپولوژیهای نادرست و تفسیرهای تکاملی گمراهکننده شود.
۳. محدودیتهای مدل (Model Limitations)
بسیاری از روشهای فیلوژنتیک بر مدلهای صریح تکامل تکیه دارند، که تسهیلهای سادهشدهای از فرآیندهای پیچیده زیستی ایجاد تنوع هستند. روشهای مبتنی بر فاصله فرض میکنند که تفاوتهای جفتی بهطور کافی روابط تکاملی را بازتاب میدهند. روشهای حداکثر احتمال (Maximum Likelihood) و بیزی (Bayesian) از مدلهای احتمالاتی جایگزینی استفاده میکنند که نرخ تغییرات میان شخصیتها یا توالیها را توصیف میکنند.
با این حال، هیچ مدلی واقعیت را بهطور کامل بازنمایی نمیکند. نرخهای جایگزینی ممکن است در شاخهها و موقعیتها متفاوت باشد، فرآیندهای تکاملی ممکن است غیرایستگاه (non-stationary) باشند و تعاملات میان ژنها یا شخصیتها ممکن است فرضیات مدل را نقض کنند. برای مثال، مدلهای ساعت مولکولی فرض میکنند که نرخها در طول زمان تقریبا ثابت هستند، اما ناهمگنی نرخها رایج است، بهویژه در ژنهای به سرعت تکاملیابنده یا بسیار محدود. اشتباه در مشخص کردن مدل میتواند منجر به توپولوژیهای سوگیر، طول شاخههای نادرست و نتیجهگیریهای گمراهکننده شود. انتخاب دقیق مدل، آزمایش مدلهای جایگزین و استفاده از مدلهای پیچیده یا شلشده میتواند این مسائل را کاهش دهد، اما آنها را بهطور کامل رفع نمیکند.
۴. اختلاف بین درخت ژن و درخت گونه (Gene Tree–Species Tree Discordance)
یکی از محدودیتهای زیستی مهم ناشی از اختلاف بین درخت ژن و درخت گونه است. ژنها یا لوکوسهای فردی ممکن است تاریخچه تکاملی متفاوتی از تاریخچه کلی گونهها داشته باشند، به دلیل فرآیندهایی مانند تکثیر ژن، انتقال افقی ژن، جداسازی ناقص شجرهای (incomplete lineage sorting) یا بازترکیبی. این اختلاف بهویژه در تکامل میکروبی، گیاهان پلیپلوئید و شاخههای اخیراً جداشده آشکار است.
برای مثال، در باکتریها، انتقال افقی ژن میتواند منجر به ایجاد درخت ژنی شود که گونههای غیرمرتبط را گروهبندی میکند، در حالی که درخت گونه روابط کلی موجودات را منعکس میکند. به همین ترتیب، در گروههای مهرهدار با تابش سریع، جداسازی ناقص شجرهای میتواند درختهای ژنی متناقضی ایجاد کند که جدایی واقعی در سطح گونه را مخدوش میکند. رفع این اختلاف نیازمند تحلیل دقیق با استفاده از روشهای مبتنی بر کوآلسنت، دادههای چند لوکوس و رویکردهای آماری است که فرآیندهای تصادفی تکامل ژن را مدلسازی میکنند.
۵. جاذبه شاخه بلند و ناهمگنی نرخ (Long-Branch Attraction and Rate Heterogeneity)
جاذبه شاخه بلند (LBA) یک خطای شناختهشده در فیلوژنتیک است، بهویژه در روشهای پارسیمونی و برخی روشهای مبتنی بر فاصله. LBA زمانی رخ میدهد که شاخههای به سرعت در حال تکامل به طور مصنوعی نزدیک به هم به نظر برسند به دلیل تجمع تغییرات متعدد در طول شاخههای بلند. این پدیده میتواند استنتاج را گمراه کند و تاکساهای دور از هم را گروهبندی کند و خویشاوندان نزدیک را حذف کند.
ناهمگنی نرخ در طول سایتها و شاخهها این مشکل را تشدید میکند. موقعیتهای بسیار متغیر در توالی DNA یا پروتئین ممکن است سریع اشباع شوند و سیگنال فیلوژنتیک را از دست بدهند، در حالی که موقعیتهای بهطور آهسته تکاملیابنده ممکن است وضوح کافی برای جداییهای اخیر را نداشته باشند. روشهای مبتنی بر مدل با پارامترهای ناهمگنی نرخ، مانند نرخهای توزیعیافته گاما، و تراز دقیق توالیها میتوانند LBA را کاهش دهند، اما این چالش به ویژه در تحلیلهای تکامل عمیق باقی میماند.
۶. انتقال افقی ژن و تکامل شبکهای (Horizontal Gene Transfer and Reticulate Evolution)
در حالی که اکثر درختهای فیلوژنتیک ساختار دوتایی شاخهای را فرض میکنند، تکامل شبکهای شامل انتقال افقی ژن، هیبریداسیون و بازترکیبی است که روابط شبکهای ایجاد میکند و نمیتواند به طور کامل توسط درختهای سنتی بازنمایی شود. این امر بهویژه در پروکاریوتها، جایی که تبادل ژن رایج است، و گیاهان، جایی که رویدادهای هیبریداسیون خطهای پیچیده ایجاد میکنند، رایج است.
روشهای استاندارد ساخت درخت ممکن است نتوانند رویدادهای شبکهای را بهدرستی نشان دهند، توپولوژیهای گمراهکننده ایجاد کنند یا سیگنالهای تکاملی مهم را کاهش دهند. شبکههای فیلوژنتیک، تجزیه تقسیم (split decomposition) و سایر روشهای تخصصی برای این فرآیندها توسعه یافتهاند، اما تفسیر آنها نیازمند توجه دقیق به زمینه زیستی و محدودیتهای داده است.
۷. مشکلات همترازی و کیفیت دادهها (Alignment and Data Quality Issues)
استنتاج دقیق فیلوژنتیک وابسته به همترازی توالیهای با کیفیت بالا است. توالیهای ناهمتراز، گنجاندن ژنهای غیر اورتوگ یا خطاهای توالییابی میتوانند سیگنال تکاملی را تحریف کنند. Indelها، نواحی مبهم و توالیهای تکراری چالشهای اضافی ایجاد میکنند، به ویژه در دادههای بزرگ یا بسیار متنوع.
پژوهشگران باید دادهها را با دقت بررسی، نواحی نامناسب همترازی را حذف و اورتولوژی را تأیید کنند تا از ایجاد خطا جلوگیری شود. نرمافزارهای همترازی چندتایی، بازبینی دستی و ملاحظات ساختاری برای اطمینان از بازتاب دقیق همولوژی توالیها استفاده میشوند. خطاها در همترازی مستقیماً به استنتاج درخت منتقل میشوند، که اهمیت آمادهسازی دقیق دادهها را نشان میدهد.
۸. محدودیتهای محاسباتی (Computational Limitations)
ساخت درختها، به ویژه با روشهای مبتنی بر مدل و دادههای بزرگ، میتواند شدیداً نیازمند منابع محاسباتی باشد. روشهای حداکثر احتمال و بیزی نیاز به ارزیابی تعداد زیادی توپولوژی ممکن دارند که با افزایش تعداد تاکساها به صورت فاکتوریلی رشد میکند. دادههای در مقیاس ژنوم ممکن است شامل هزاران توالی و میلیونها نوکلئوتید باشند، که حتی برای محیطهای محاسباتی با عملکرد بالا نیز چالش ایجاد میکند.
الگوریتمهای هیواریستیک، محاسبات موازی و روشهای تقریبی تحلیل دادههای بزرگ را ممکن ساختهاند، اما محدودیتهای محاسباتی همچنان مانع جستجوی جامع و برازش مدلهای پیچیده میشوند. پژوهشگران باید اندازه داده، پیچیدگی مدل و منابع محاسباتی را متعادل کنند و اغلب بین سرعت و دقت تصمیمگیری کنند.
۹. چالشهای تفسیر (Interpretational Challenges)
حتی زمانی که درختها به درستی استنتاج شدهاند، تفسیر آنها نیازمند توجه دقیق است. درختها فرضیههایی از تاریخچه تکاملی هستند، نه بازنماییهای مطلق حقیقت. طول شاخه، توپولوژی و مقادیر حمایت همگی عدم قطعیت را حمل میکنند، و نادیده گرفتن این عوامل میتواند منجر به تفسیر نادرست شود. برای مثال، وجود یک شاخه کوتاه لزوماً به معنای جدایی اخیر نیست؛ ممکن است نشاندهنده دادههای محدود یا نرخهای تکامل کند باشد.
بهطور مشابه، گنجاندن تنها یک زیرمجموعه از تاکساها میتواند توپولوژی درخت را تحریف کرده و گروهبندیهای مصنوعی ایجاد کند. پژوهشگران باید همیشه محدودیتهای داده، روش و مدل خود را در نظر بگیرند و نتایج را با احتیاط و شفافیت ارائه دهند.
بخش نهم: مسیرهای آینده در پژوهشهای فیلوژنتیک
پژوهشهای فیلوژنتیک طی چند دهه گذشته به سرعت پیشرفت کردهاند، و مسیر خود را از تحلیل شخصیتهای مورفولوژیکی به سمت رویکردهای مولکولی و در مقیاس ژنوم تغییر دادهاند. با وجود این پیشرفتها، این حوزه همچنان در حال تحول است و ظهور فناوریهای نوین، روشهای محاسباتی و چارچوبهای مفهومی جدید مسیر آینده آن را شکل میدهد. مسیرهای آینده در پژوهشهای فیلوژنتیک تحت تأثیر افزایش دسترسی به دادههای ژنومی، پیشرفت در مدلسازی آماری، ادغام با سایر رشتههای زیستی و نیاز به پرداختن به فرآیندهای تکاملی پیچیده قرار دارند. این تحولات وعده میدهند که درک ما از درخت زندگی را بهبود بخشند، دقت استنتاج فیلوژنتیک را افزایش دهند و کاربردهای تحلیلهای تکاملی را در زیستشناسی و جامعه گستردهتر کنند.
۱. ادغام دادههای ژنومی و چند-اُمیک (Integration of Genomic and Multi-Omic Data)
یکی از مسیرهای امیدوارکننده در پژوهشهای فیلوژنتیک، ادغام دادههای چند-اُمیک است، از جمله ژنومیک، ترنسکریپتومیک، پروتئومیک، متابولومیک و اپیژنتیک. توالییابی ژنوم کامل قبلاً تحول بزرگی در فیلوژنتیک ایجاد کرده است و امکان تحلیلهای گسترده ژنومی برای روابط تکاملی را فراهم کرده است.
ادغام این دادههای متنوع میتواند دید جامعتری از فرآیندهای تکاملی ارائه دهد، زیرا اطلاعاتی درباره بیان ژن، عملکرد پروتئین، مسیرهای متابولیک و تنظیمات اپیژنتیکی را وارد تحلیل میکند.
برای مثال، ترنسکریپتومیک مقایسهای میتواند نشان دهد که الگوهای بیان ژن چگونه در میان شاخهها متفاوت شدهاند و پیامدهای عملکردی تغییرات تکاملی را روشن کند. پروتئومیک اطلاعاتی درباره توالیها، ساختارها و تعاملات پروتئینهای حفظشده و متنوع ارائه میدهد که میتواند استنتاج فیلوژنتیک و تکامل عملکردی را تقویت کند. ادغام دادههای چند-اُمیک نیازمند ابزارهای محاسباتی پیشرفته برای مدیریت انواع دادههای مختلف است، اما پتانسیل آشکار کردن الگوهای تکاملی پیچیده که در تحلیلهای تکدادهای پنهان ماندهاند را دارد.
۲. فیلوژنومیک و رویکردهای دادهکلان (Phylogenomics and Big Data Approaches)
گسترش مداوم توالییابی ژنوم در تاکساهای متنوع امکان توسعه فیلوژنومیک را فراهم کرده است، روشی که صدها تا هزاران لوکوس را بهطور همزمان تحلیل میکند. این رویکرد توان آماری و وضوح درختهای فیلوژنتیک را به ویژه برای تابشهای سریع، انشعابات عمیق و گونههای نزدیک به هم افزایش میدهد. با کاهش هزینههای توالییابی و بهبود فناوریهای با توان بالا، تعداد ژنومهای موجود به صورت نمایی رشد خواهد کرد و فرصتهای تحلیلهای جامعتر را ایجاد میکند.
رویکردهای دادهکلان در فیلوژنومیک نیازمند توسعه الگوریتمهای مقیاسپذیر و راهکارهای محاسباتی با عملکرد بالا هستند. یادگیری ماشین و هوش مصنوعی بهتدریج در استنتاج فیلوژنتیک به کار گرفته میشوند و امکان شناسایی خودکار ژنهای اورتوگ، بهبود همترازی و شناسایی الگوهای تکاملی را فراهم میکنند. این رویکردها میتوانند دقت درخت را افزایش دهند، سیگنالهای انتخاب را شناسایی کنند و فرآیندهای پیچیده تکاملی مانند هیبریداسیون، انتقال افقی ژن یا تکثیر ژن را آشکار کنند.
۳. پرداختن به تکامل شبکهای و شبکههای فیلوژنتیک (Addressing Reticulate Evolution and Networks)
یکی از مرزهای اصلی پژوهش فیلوژنتیک مربوط به تحلیل تکامل شبکهای است، شامل هیبریداسیون، انتقال افقی ژن، اینتروگرشن و بازترکیبی. درختهای شاخهای سنتی نمیتوانند این فرآیندها را بهدرستی بازنمایی کنند، و نیازمند استفاده از شبکههای فیلوژنتیک و مدلهای مبتنی بر گراف هستیم. این روشها اجازه میدهند رویدادهای تکاملی که الگوی شاخهای ساده ندارند را تجسم و کمیسازی کنیم و بازنمایی واقعیتری از تاریخچه موجودات ارائه دهیم.
برای مثال، در گیاهان و میکروبها، هیبریداسیون و انتقال افقی ژن بسیار رایج است، که تاریخچههای تکاملی پیچیدهای ایجاد میکنند که توسط درختهای استاندارد قابل مشاهده نیستند. روشهای شبکهای همراه با دادههای ژنومی و ترنسکریپتومی امکان بازسازی تاریخچههای شبکهای و شناسایی شاخههای دهنده و گیرنده را فراهم میکنند. پژوهشهای آینده احتمالاً گسترش کاربرد روشهای مبتنی بر شبکه، توسعه چارچوبهای آماری قوی و ابزارهای محاسباتی برای مدیریت دادههای بزرگ و پیچیده را دنبال خواهند کرد.
۴. توسعه مدلهای تکامل بهبود یافته (Improved Models of Evolution)
یکی دیگر از حوزههای حیاتی برای پژوهشهای فیلوژنتیک آینده، توسعه مدلهای پیشرفته برای تکامل توالی و ویژگیها است. مدلهای فعلی اغلب فرضیات سادهای درباره نرخهای جایگزینی، استقلال سایتها و یکنواختی در طول شاخهها دارند. با این حال، فرآیندهای واقعی تکاملی متنوع، وابسته به زمینه و تحت تأثیر انتخاب، رانش ژنتیکی، بازترکیبی و اپیستازی هستند.
مدلهای آینده احتمالاً این پیچیدگیها را وارد خواهند کرد، امکان نرخهای تکاملی ویژه هر سایت، تغییر نرخ در طول شاخهها، انتخاب در سطح کدون و محدودیتهای ساختاری یا عملکردی را فراهم میکنند. پیشرفت در مدلسازی احتمالاتی و آمار محاسباتی اجازه میدهد مدلهای پیچیده به دادههای بزرگ تطبیق داده شوند، دقت استنتاج فیلوژنتیک و تخمین طول شاخه را افزایش دهند. مدلهای بهبود یافته همچنین زمانبندی دقیقتر انشعابات، بازسازی توالیهای اجدادی و شناسایی تکامل تطبیقی را امکانپذیر میکنند.
۵. ادغام دادههای فسیلی و تاریخگذاری نوکها (Integration with Fossil Data and Tip-Dating)
ادغام شواهد فسیلی در تحلیلهای فیلوژنتیک همچنان یک چالش بزرگ اما فرصت حیاتی است. روشهای Tip-dating و Total-evidence دادههای مورفولوژیکی فسیلها را با توالیهای مولکولی تاکساهای زنده ترکیب میکنند تا زمان انشعاب و بازسازی تاریخچه تکاملی را تخمین بزنند. توسعههای آینده احتمالاً ادغام دادههای فسیلی، مدلهای بهبود یافته برای شخصیتهای گمشده، سوگیریهای حفظ و تکامل مورفولوژیکی را بهبود میبخشند.
تصویربرداری با وضوح بالا، بازسازی سهبعدی و دیرینهشناسی مجازی امکان تحلیل مورفولوژیکی دقیق فسیلها را فراهم میکنند. ترکیب این فناوریها با روشهای محاسباتی پیشرفته اجازه میدهد شاخههای منقرض شده را در درختهای فیلوژنومیک وارد کنیم و دید دقیقتر و غنیتری از الگوهای ماکروتکاملی و زمانبندی نوآوریهای کلیدی داشته باشیم.
۶. فیلودینامیک و ردیابی تکاملی در زمان واقعی (Phylodynamics and Real-Time Evolutionary Tracking)
روشهای فیلوژنتیک بهطور فزایندهای برای ردیابی تکاملی در زمان واقعی، به ویژه در اپیدمیولوژی، اکولوژی و حفاظت به کار میروند. فیلودینامیک ترکیبی از استنتاج فیلوژنتیک، ژنتیک جمعیت، اپیدمیولوژی و مدلسازی اکولوژیکی است تا دینامیک جمعیتهای به سرعت تکاملیابنده را درک کند.
برای مثال، در تکامل ویروسها، توالییابی در زمان واقعی و تحلیل فیلوژنتیک میتواند ظهور واریانتها را ردیابی، مسیرهای انتقال را پیشبینی و پاسخهای بهداشت عمومی را هدایت کند. رویکردهای مشابه در اکولوژی میکروبی، پایش گونههای مهاجم و ژنتیک حفاظتی برای ردیابی ساختار جمعیت، جریان ژن و تکامل تطبیقی اعمال میشوند. پژوهشهای آینده روشهای فیلودینامیک را گسترش داده و ترکیب توالییابی با توان بالا، پایش محیطی و مدلسازی محاسباتی امکان تحلیل سریع و پیشبینی دینامیک تکاملی در شرایط واقعی را فراهم میکنند.
۷. کاربردها در زیستشناسی مصنوعی و مهندسی تکاملی (Applications in Synthetic Biology and Evolutionary Engineering)
تحلیل فیلوژنتیک همچنین در زیستشناسی مصنوعی و مهندسی تکاملی کاربردهای امیدوارکننده دارد. درک تاریخچه تکاملی ژنها، پروتئینها و مسیرها به پژوهشگران کمک میکند طراحی بیومولکولهای جدید، بهینهسازی شبکههای متابولیک و مهندسی موجودات با ویژگیهای دلخواه را انجام دهند.
برای مثال، آزمایشهای تکامل هدایتشده اغلب بر اساس بازسازی توالی اجدادی با راهنمایی درختها انجام میشوند تا آنزیمهایی با پایداری، فعالیت یا اختصاصیت بستر افزایش یافته تولید کنند. همچنین، بینشهای فیلوژنتیک میتوانند طراحی مدارهای ژنی مصنوعی، کاهش ناپایداری تکاملی و پیشبینی تعاملات با میزبان را راهنمایی کنند. ادغام آینده فیلوژنتیک با زیستشناسی مصنوعی قدرت پیشبینی طراحی تکاملی را گسترش داده و مهندسی منطقی سیستمهای زیستی را امکانپذیر میکند.
۸. دموکراتیزه کردن ابزارهای فیلوژنتیک و آموزش (Democratization of Phylogenetic Tools and Education)
با پیچیدهتر شدن روشهای فیلوژنتیک، تاکید بر دسترسی آسانتر به ابزارها برای پژوهشگران، مربیان و دانشجویان افزایش یافته است. نرمافزارهای کاربرپسند، پلتفرمهای تحت وب و ابزارهای تصویری تعاملی در دسترس قرار گرفتهاند تا درگیری گستردهتری با تحلیل تکاملی ایجاد شود.
مسیرهای آینده شامل محاسبات ابری، نمایش تعاملی درختها و ادغام با منابع آموزشی است. با کاهش موانع ورود، این توسعهها پژوهشهای بینرشتهای را تسهیل، سواد علمی را افزایش و استفاده از بینشهای فیلوژنتیک در حوزههای کاربردی مانند پزشکی، کشاورزی و حفاظت را ارتقا میدهند.